
Глава 3 Физико-химические методы анализа








3.1. Классификация методов ФХМА
Физико-химические методы анализа (ФХМА) анализа представлены электрохимическими, оптическими, хроматографическими и другими приборными методами.
К группе электрохимических методов относятся:
Потенциометрия - метод основан на измерении электродвижущей силы (ЭДС) электродвижущего элемента.
Вольтамерометрия - метод анализа, основанный на расшифровке поляризационных кривых (вольтамперограмм), получаемых в электролитической ячейке с поляризующимся индикаторным электродом и неполяризующимся электродом сравнения.
Кондуктометрия – метод основан на измерение электрической проводимости с ростом концентрации.
Кулонометрия – в кулонометрических методах определяют количество электричества, которое расходуется в ходе электрохимической реакции.
Термометрическое титрование – основано на измерении теплового эффекта реакции титрования или величин пропорциональных этому тепловому эффекту. Существуют и другие методы.
Оптические методы анализа основаны на измерении показателей оптических свойств анализируемых веществ.
К группе оптических методов анализа относятся следующие методы:
Абсорбционная спектроскопия - основана на измерении количества света, поглощенного исследуемым раствором, и изучении спектров поглощения вещества, являющихся его индивидуальной характеристикой. Различают фотоколориметрический и спектрофотометрический методы.
Фотоколориметрический метод основан на определении спектра поглощения или измерении светопоглощения в видимой части спектра. При этом в качестве источника света применяют или свет обычной лампы накаливания ("белый" свет) или же "белый" свет, пропущенный предварительно через широкополосные светофильтры.
Спектрофотометрический метод основан на определении спектров поглощения или измерении светопоглощения (в ультрафиолетовой, видимой и инфракрасной областях спектра) при строго определенной длине волны (монохроматическое излучение).
Нефелометрия - основана на измерении интенсивности света, отраженного или рассеянного суспензией определяемого вещества.
Турбидиметрия - основана на измерении интенсивности света, прошедшего через мутный раствор, содержащий частицы определяемого вещества.
Люминесцентный анализ - основан на измерении интенсивности света, излучаемого веществом вследствие люминесценции.
Эмиссионный спектральный анализ - это физический метод определения химического состава вещества по его спектру, испускаемому возбужденными атомами или молекулами (например, электрической дугой, высоковольтной искрой).
Рефрактометрия - основана на измерении коэффициента преломления света определяемым веществом.
Поляриметрия - основана на изучении вращения плоскости поляризации света анализируемым веществом.
Хроматографические методы анализа, или хроматогра́фия (от др. -греч. χρῶμα — «цвет») — метод разделения и анализа смесей веществ, а также изучения физико-химических свойств веществ. Основан на распределении веществ между двумя фазами — неподвижной (твёрдая фаза или жидкость, связанная на инертном носителе) и подвижной (газовая или жидкая фаза, элюент).
По агрегатному состоянию фаз хроматографические методы анализа делятся на:
- газожидкостные, где подвижной фазой служит поток инертного газа, который проходит через жидкий сорбент.
- Жидкостно‑жидкостные, где в качестве элюента и неподвижной фазы используются жидкие среды, и др.
- газовые, где в качестве элюента и неподвижной фазы используются газовые среды.
Для выполнения большинства методов необходимо хроматографическое оборудование, чаще всего колоночный хроматограф: адсорбция осуществляется в колонках, заполненных неподвижной фазой. Но иногда используется плоскостная хроматография, в которой используется тонкий срез сорбента или специальная бумага. Также в последнее время получили распространение капиллярный хроматографический метод, при котором разделение происходит в пленке жидкости, и хроматография в полях, требующая для проведения анализа создания дополнительных магнитных, центробежных или иных сил.
Хроматография как способ разделения сложных смесей в сочетании с другими методами анализа универсальный метод, позволяющий определять фальсификацию сливочного масла, отличать "вологодское масло" от подделки, определять процент соевого белка в продукте, выявлять применение запрещённых красителей, определять количественно витамины, биоэлементы, тяжёлые металлы, остаточные пестициды в почве, кормах и др.
Высокоэффективная жидкостная хроматография (ВЭЖХ), называемая также жидкостной хроматографией высокого давления, – наиболее перспективный аналитический вариант классической колоночной хроматографии в современном приборном исполнении. ВЭЖХ позволяет проводить одновременное разделение сложных проб на составляющие их компоненты и их качественное или количественное определение.
3.2. Электрохимические методы анализа
Классификация электрохимических методов химического анализа в зависимости от измеряемого сигнала (измеряемый сигнал -метод анализа, используемый прибор): потенциал - потенциометрия, рН-метр, ионометрия, иономер-универсальный; количество электричества (в Кулонах) - кулонометрия, кулонометр; электропроводность - (от англ. conductivity -электропроводность - кондуктометрия, кондуктометр; зависимость тока (в Амперах) от напряжения (в Вольтах) - вольтамперометрия, вольтаметометр, комплексы аналитические вольтамперометрические.
3.2.1. Потенциометрический анализ
Потенциометрия – раздел аналитической химии, в котором концентрацию определяемого иона находят по величине электродвижущей силы (ЭДС) гальванического элемента.
ЭДС = Е1 – E2 (3.1);
где: Е1 и Е2 – потенциалы двух типов электродов измерительного и электрода сравнения.
Электродный потенциал в потенциометрии рассчитывается по уравнению Нернста 3.2.
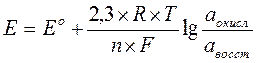 (3.2);
(3.2);
где: Е° – стандартный электродный потенциал, В;
R – универсальная газовая постоянная, 8,314 Дж/град×моль;
Т – абсолютная температура, Т;
n – число электронов, участвующих в данной электродной реакции;
F – число Фарадея, 96500 К;
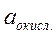 – активность в растворе окисленной формы;
– активность в растворе окисленной формы;
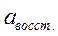 . – активность в растворе восстановленной формы.
. – активность в растворе восстановленной формы.
При температуре 25С° 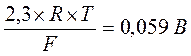 , и уравнение Нернста можно записать в следующем виде, уравнение 3.3.
, и уравнение Нернста можно записать в следующем виде, уравнение 3.3.
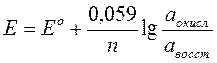 (3.3)
(3.3)
Уравнение Нернста выражает зависимость электродного потенциала от концентрации определяемого иона:
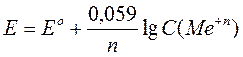 (3.4),
(3.4),
где: Е – определяемый потенциал, В;
Е° – стандартный (нормальный) электродный потенциал, В;
n – число электронов, участвующих в данной электродной реакции;
С – концентрация определяемого иона, моль/л.
рН–метр – ( иономер, милливольтметр) предназначен для измерения активности ионов водорода (рН), концентрации ионов в соответствии с используемым селективным электродом, окислительно-восстановительных потенциалов (Еh) и температуры водных растворов. Измерение рН, Еh и температуры осуществляется в цифровой форме с помощью измерительного преобразователя и набора электродов.
В основу работы рН-метра положен потенциометрический метод измерения рН и Еh контролируемого раствора.
При измерении рН (или Е h) растворов используется система, состоящая из измерительного или индикаторного и вспомогательного или электрода сравнения электродов.
В качестве измерительного электрода при измерении рН используется стеклянный электрод, а в качестве вспомогательного – хлорсеребряный электрод (рис.9.).
Электродная система при погружении в контролируемый раствор развивает ЭДС, линейно зависящую от активности ионов и температуры раствора. Контакт вспомогательного электрода с контролируемым раствором осуществляется с помощью электролитического ключа, обеспечивающего истечение насыщенного раствора KCl в контролируемый раствор.
Раствор хлорида калия непрерывно просачивается через электролитический ключ, предотвращая проникновение из контролируемого раствора в систему вспомогательного электрода посторонних ионов, которые могли бы изменить величину потенциала электрода, ЭДС электродной системы преобразуется и считывается с индикатора рН-метра.
При измерении окислительно-восстановительного потенциала в качестве измерительного электрода используется редоксметрический электрод, в качестве вспомогательного – хлорсеребряный электрод. Измерение Eh производится в мВ (рис.3.1.).
|
|
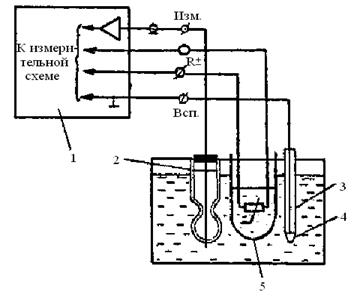
Рис. 3.1. Схема подключения электродной системы при измерении рН.
1 – преобразователь; 2 – измерительный стеклянный электрод; 3 – вспомогательный электрод; 4 – электрический ключ; 5 – автоматический термокомпенсатор.
Перед работой на рН-метре его необходимо градуировать. В качестве градуировочных растворов рекомендуется применять эталоны буферных растворов pH: 1,65, 4,01, 6,86, 9,18, 12,43 при 25 ºC по ГОСТ 8.135-2004. В качестве первого градуировочного желательно использовать раствор, с pH, наиболее близким к значению pHi, второго – наиболее отдаленный от значения pHi, но находящийся в диапазоне предполагаемых измерений. Разница в значениях pH у растворов должна быть не менее единицы. Температура применяемых при градуировке прибора градуировочных растворов должна быть одинаковой (±1,5 о C). Погрешность измерений меньше, если температура градуировочных растворов близка к температуре анализируемых растворов. При использовании градуировочных растворов имеющих температуру окружающей среды рекомендуется выдержать их при комнатной температуре не менее часа. Растворы с другой температурой термостатируют.
3.2.1.1. Определение рН растворов кислот, оснований и гидролизующихся солей на иономере ЭВ-74 или на рН-150М
Познакомьтесь с прибором ЭВ-74, рН-150М или иной маркой рН-метра (иономера) по паспорту прибора.
Основные химическая посуда, реактивы:
Химические стаканчики для образцов; промывалка с дистиллированной водой; фильтровальная бумага.
Ход работы:
1) Знакомство с прибором. Например, иономер ЭВ-74 состоит из преобразователя, подставки для крепления электродов и установки сосудов с раствором.
Шкала прибора имеет следующие оцифровки:
- 1.. .19 – для измерения рН раствора на широких диапазонах.
- 1...4, 4...9, 9...14, 14...19 – для измерения рН в соответствующих диапазонах.
Вид определяемого иона (х) соответствует типу применяемого электрода.
Органы оперативного управления: тумблер "сеть"; ручки регистров "калибровка", "крутизна", "температура раствора"; кнопки выбора рода работы, кнопки выбора диапазона измерений. Регистры "калибровка" и "крутизна" служат для настройки прибора.
2) Включение прибора. Перед включением прибора в сеть необходимо:
- нажать кнопку "t".
- переключатель "пределы измерений" поставить в положение "-1...19".
- проверить подключение электродов; стеклянный электрод должен быть подключен в гнездо "изм", хлорсеребряный электрод - в гнездо "всп", термокомпенсатор – в гнездо "термокомпенсатор".
3) Настройка прибора по буферным растворам:
- Электроды перед погружением в буферный раствор тщательно промыть дистиллированной водой, остатки воды удалить фильтровальной бумагой.
- Электроды поместить в буферный раствор со значением рН = I,68 на глубину 2...4 см.
- Нажать кнопки "катион-анион" и "рХ".
- Рукояткой "калибровка" установить стрелку на значение 1,68 по шкале "-1...19".
- Включить диапазон измерений "-1...4". Если показание прибора не соответствует значению l,68, то установить стрелку рукояткой "калибровка" на значение l,68 по шкале "-1...4".
- Нажать кнопки "t" и "-1...19", снять стаканчик с буферным раствором, промыть и подсушить электроды.
- Погрузить электроды в буферный раствор со значением рН = 9,18.
- Нажать кнопку "рХ".
- Рукояткой "крутизна" установить стрелку на значение 9,18 по шкале "-1...19".
- Включить диапазон измерений "9...14". Если показание прибора не соответствует значению 9,18, то установить стрелку рукояткой "крутизна" на значение 9,18 по шкале "9. ..14".
- Нажать кнопки "t" и "-1...19".
Настройку повторить 2...3 раза. Электроды промыть и погрузить в стаканчик с дистиллированной водой.
4) Измерение рН растворов:
- анализируемый раствор ( если известна, концентрация раствора):
- показание прибора по шкале "-1…19", выбранный диапазон измерений;
- стаканчик налить один из анализируемых растворов;
- поместить электроды в анализируемый раствор;
- нажать кнопку "рХ";
- измерить рН по шкале "-1...19";
- выбрать диапазон точного измерения;
- измерить рН в выбранном диапазоне и записать результат измерений;
- нажать кнопки "t" и "-1...19";
- промыть и подсушить фильтровальное бумагой электроды.
- вылить анализируемый раствор в склянку, стаканчик промыть дистиллированной водой;
- значение рН анализируемого раствора, значение рН анализируемого раствора, рассчитанный теоретически;
- в таком же порядке измерить рН всех анализируемых растворов.
3.2.1.2 Определение рН молока и молочных продуктов
Выполнение работы:
- включить прибор рН-метр в сеть и настроить по буферным растворам (работу 3.2.1.1);
- электроды опустить в стаканчик с молоком, снять показания;
- обмыть электроды из промывалки над стаканом;
- электроды после молока протереть ватой со спиртом и далее снова обмыть водой;
- просушить электроды фильтровальной бумагой.
3.2.1.3 Определение рН образцов почв
Повышенная кислотность почв вредна для большинства сельскохозяйственных культур. Под действием ее гибнут в почве полезные микроорганизмы (клубеньковые бактерии, азотобактерии и др.). Проверяя кислотность, определяют необходимость ее известкования.
Различают две формы почвенной кислотности: актуальную и потенциальную (обменную). Актуальная кислотность обусловлена наличием свободных ионов водорода в почвенном растворе. Для определения актуальной кислотности готовят водную вытяжку из почвы и измеряют ее рН. Обменная кислотность обусловлена присутствием в почве ионов водорода и алюминия. Для определения обменной кислотности готовят солевую вытяжку и измеряют рН.
Ход работы:
1. На технохимических весах взвесить 20 г почвы.
2. Добавить 50 мл 1 н раствора хлорида калия.
3. Перемешать содержимое стаканчика в течение 5 минут и оставить на 30...60 минут.
4. Содержимое стаканчика перемешать и измерить рН.
В зависимости от значения рН солевой вытяжки почвы считают:
| сильнокислыми | рН = 3...4, |
| кислыми | рН = 4...5, |
| слабокислыми | рН = 4.. .6, |
| нейтральными | рН = 7, |
| слабощелочными | рН = 7.. .8, |
| щелочными | рН = 8…9 |
| сильнощелочными | рН = 9. . .1 1. |
Определите характер анализируемого образца почвы. Почвы с рН солевой вытяжки меньше 4,5 нуждаются в известковании.
3.2.2 Раздельное определение компонентов в бинарных смесях с помощью автоматического титрования
Кислотно-основное автоматическое титрование выполняют автоматическом режиме на автотитраторах типа БАТ-15.2МП или аналогичного.
Для изучения методов анализа одновременно двух компонентов в пробах, используя метод кислотно-основного потенциометрического автоматического титрования (автоматического рН-метрического титрования) используем пробы кислот и кислых солей А и Б:
- проба А – смесь H3РO4 и NaH2РO4;
- проба Б – смесь H3РO4 и H2SO4.
Сущность работы. Потенциометрическая индикация конечной точки титрования (к.т.т.) позволяет дифференцированно титровать смеси кислот с погрешностью до 0,1%, если Ka,1 / Ka,2 ≥ 104 , при этом константа диссоциации слабой кислоты должна быть не ниже 10–8 (pKа < 8).
В качестве индикаторного электрода применяют стеклянный электрод, а в качестве электрода сравнения – хлоридсеребряный. В современных моделях иономеров в автотитраторах эти электроды объединены в один комбинированный электрод (датчик).
Проба А. Смесь H3РO4 и NaH2РO4.
При титровании смеси H3РO4 и NaH2РO4 щелочью на кривой титрования наблюдается 2 скачка.
Первый из них отвечает оттитровыванию H3PO4 по первой ступени: H3PO4 + NaOH = NaH2PO4 * + H2O (3.1)
Второй скачок соответствует оттитровыванию H3PO4 по второй ступени и соли NaH2РO4, которая содержалась в анализируемой пробе:
NaH2PO4 * + NaOH = Na2НPO4 + H2O (3.2)
NaH2PO4 + NaOH = Na2НPO4 + H2O (3.3)
Проба Б. Смесь H3РO4 и H2SO4.
При титровании смеси кислот H2SO4 и H3PO4 щелочью на кривой титрования наблюдается 2 скачка.
Первый из них отвечает оттитровыванию всей H2SO4, а также H3PO4 по первой ступени:
H2SO4 + 2NaOH = Na2SO4 + 2H2O; (3.4)
H3PO4 + NaOH = NaH2PO4 + H2O. (3.5)
Второй скачок соответствует оттитровыванию H3PO4 по второй ступени:
NaH2PO4 + NaOH = Na2НPO4 + H2O (3.6)
Основные химическая посуда, реактивы:
блок автоматического титрования БАТ с рН-метром или иономером; индикаторный электрод – стеклянный; электрод сравнения – хлоридсеребряный (или один комбинированный электрод); магнитная мешалка со стержнем; 0,1 M стандартный раствор NaOH или KОН; бюретка; стакан для титрования вместимостью 150 мл.
Ход работы:
Получают анализируемый раствор в стакан для титрования и разбавляют водой до погружения электродов. Включают магнитную мешалку. Затем проводят титрование, добавляя щелочь по 0,2–0,5 мл и фиксируя значение рН после добавления каждой порции титранта. Титрование прекращают после второго скачка, когда значение рН раствора практически не меняется. Строят интегральные (рН – V, мл) и дифференциальные (ΔрН / ΔV – V, мл) кривые титрования.
По ним определяют объемы титранта, необходимые для достижения первой и второй к. т. т. Используя полученные значения, находят массу (г) каждого компонента в выданной для анализа пробе. При титровании с использованием БАТа предварительно рассчитывают значения рН раствора в первой и второй точках эквивалентности с целью задания их для автоматического титрования. Поскольку в первой точке эквивалентности в растворе в обоих случаях присутствует амфолит NaH2PO4, то расчет рН ведут по формуле 3.7.
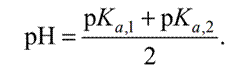
| (3.7.) |
Во второй точке эквивалентности в растворе в обоих случаях присутствует амфолит Na2HPO4, следовательно,
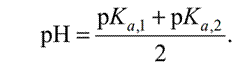
| (3.8.) |
Необходимо предварительно определить объемы титранта, которые израсходованы на каждую из протекающих реакций по отдельности:
Проба А – на титрование H3PO4 по первой ступени (реакция 2.1) затрачен объем щелочи V1, следовательно, на титрование H3PO4 по второй ступени (реакция 2.2) затрачен точно такой же объем щелочи V1.
Тогда на титрование соли NaH2РO4, которая содержалась в анализируемой пробе (реакция 2.3), затрачено (V2 – 2V1) мл щелочи.
Проба Б – на титрование H3PO4 по второй ступени (реакция 3.6) затрачен объем щелочи ΔV = V2 – V1, следовательно, на титрование H3PO4 по первой ступени (реакция 3.5) затрачен точно такой же объем щелочи ΔV.
Тогда на титрование H2SO4 (реакция 3.4) затрачено (V1 – ΔV) мл щелочи.
После завершения автоматического титрования проводят расчет массы каждого компонента по закону эквивалентов, используя значения объемов титранта V1 и V2.
Метод анализа, основанный на расшифровке поляризационных кривых (вольтамперограмм), получаемых в электролитической ячейке с поляризующимся индикаторным электродом и неполяризующимся электродом сравнения, называют вольтамперометрическим. Он относится к электрохимическим методам анализа и позволяет одновременно получить качественную и количественную информацию о веществах, восстанавливающихся или окисляющихся на индикаторном электроде. Концентрация определяемого компонента (витамина, элемента, антибиотика) определяется методом добавок аттестованной смеси определяемого компонента.
Методики аттестованы в соответствии с ГОСТ Р 8.563 (ГОСТ 8.010) для следующих биологических объектов: воды (питьевые, природные, минеральные, технологически чистые, очищенные сточные); продукты пищевые и продовольственное сырьё (зерно и продукты его переработки, корма, комбикорма, комбикормовое сырьё и кормовые добавки, мука, крупа, хлеб, хлебобулочные и мукомольно-крупяные изделия; мясо и мясопродукты; молоко и молочные продукты; продукты детского питания, соки, фрукты, ягоды, витаминно-минеральные препараты, БАДы; консервированные пищевые продукты.)
3.2.3 Вольамперометрическое определение витаминов В1, В2, С в биологических объектах
Диапазон содержания в биологических объектах витамина В1 – 2 ÷20000 мг/кг; витамина В2-0,2÷5000 мг/кг; витамина С составляет 2 ÷3000 мг/кг. При более высоких концентрациях допускается разбавление подготовленной к измерению пробы или уменьшение навески анализируемой пробы.
Для выполнения работы необходимо использование вольтапмерометрического анализатора в комплекте с компьютером типа СТА-1, где регистрацию и обработку концентраций компонентов в пробе выполняет система сбора и обработки данных анализатора. Необходимы также водяная баня и лабораторная центрифуга, например, центрифуга лабораторная многофункциональная Frontier, техно-химические весы.
Основные химическая посуда, реактивы:
магнитная мешалка со стержнем; НСl с молярной концентрацией С=0,1 моль/л; сухой КСl; химические стаканы вместимостью 150 мл, бумажные фильтры
Ход работы:
Подготовка проб для определения витаминов В1 и В2
1) Навеску пробы 10-50 г, взятой с точностью 0,01 г, перенести в мерную колбу на 250 см3,
2) Добавить 100 см3 бидистиллированной воды, 2 см3 соляной кислоты с молярной концентрацией 7 моль/дм3
3) Нагревают на кипящей водяной бане в течение 30 мин. Охладить до 30÷40 ºС
4) Добавить 0,5 г хлорида марганца четырёхводного, растворить его, отцентрифугировать в течение 15 мин или отфильтровать через двойной бумажный фильтр.
5) Центрифугат сливают в колбу, к нему добавляют 3-4 г хлорида калия, осадок отфильтровывают через бумажный фильтр. Полученный фильтрат является подготовленной пробой для анализа на содержание витаминов В1 и В2.
Подготовка проб для определения витамина С
Рекомендуемые навески проб, анализируемый объект, навеска, г
1 Пюре, продукты питания на фруктовой, овощной и молочной основе, 5,0÷10,0
2 Витаминизированные препараты, 0,1÷0,5
3 Фрукты и ягоды, 5,0 ÷10,0
4 Соки, 0,1÷2,0
5 Молоко, кисломолочные напитки, 5,0 ÷50,0
6 Молочные смеси сухие, 0,2÷2,0
7 Крупы сухие, 0,1÷1,0
Навеску пробы, взятой с точностью 0,01 г, перенести в мерную колбу на 100 см3, в случае сухой пробы, предварительно измельчённой в фарфоровой ступке.
1) Для пюре овощных и фруктовых, круп сухих к пробе добавить 30 см3 НСl с молярной концентрацией С=0,1 моль/л; перемешать, довести до метки бидистиллированной водой.
2) Для фруктов и ягод пробу в мерной колбе на 100 см3 смыть бидистиллированной водой, добавить 30 см3 НСl с молярной концентрацией С=0,1 моль/л; 1 г сухого КСl, перемешать, довести до метки бидистиллированной водой.
3) Для соков навеску пробы растворить в растворе хлорида калия с молярной концентрацией С=0,1 моль/л, подкисленного НСl с молярной концентрацией С=0,1 моль/л до рН 3-4. Довести объём до метки раствором КСl с молярной концентрацией С=0,1 моль/л.
4) Для молока, кисломолочных напитков, молочных смесей сухих к навеске в колбе добавить 30 см3 бидистиллированной воды, 1 г сухого КСl, перемешать, довести до метки НСl с молярной концентрацией С=0,1 моль/л.
Доведённую до метки в колбе растворённую пробу выдержать 20-30 мин, отфильтровать через двойной бумажный фильтр. Фильтрат является подготовленной пробой для анализа на содержание витамина С.
Вольтамперометрическое (ВА) измерение подготовленной пробы
1) В кварцевый стаканчик поместить отмеренную с помощью пипетки или дозатора аликвоту полученного фильтрата 0,1÷1,0 см3 с точностью до 0,01 см3. Довести объём пробы до 10 см3 раствором фонового электролита
2) Поместить стаканчик с анализируемым раствором в электролитическую ячейку.
3) Опустить в раствор индикаторный (стеклоуглеродный) электрод и электрод сравнения (хлорсеребрянный). Подключить электроды к прибору, устанавливая потенциал -0,30 В и чувствительность прибора 4 ∙10-9…1 ∙10-9 А/мм. Включить газ азот, в течение 180 с. удалять из раствора кислород пропусканием азота.
4) Провести процесс электролиза (электронакопления) при потенциале -0,30 В в течение 30 сек при автоматическом перемешивании раствора.
5) Отключить газ и через 10 сек начать регистрацию вольамперограммы в диапазоне потенциалов от -0,30 В до +1,1 В.
6) После снятия вольтамперограммы стеклоуглеродный электрод вынимают из электрохимической ячейки, ополаскивают дистиллированной водой, опускают на 2-3 с в стаканчик с этиловым спиртом и протирают рабочую поверхность электрода фильтровальной бумагой.
7) Повторить операции последовательно 3)…6) три раза.
8) Измерить линейкой высоты анодных пиков. Если высота анодного пика витамина С превышает 200 мм, то уменьшают чувствительность прибора (загрубить), меньше 5 мм – увеличивают чувствительность прибора. После чего вновь последовательно повторяют операции 4)…6) три раза.
9) В стаканчик с анализируемым раствором с помощью пипетки или дозатора внести добавку аттестованной смеси определяемого витамина в таком объёме, чтобы высота пита на вольтамперной кривой увеличилась примерно в два раза по сравнению с первоначальной (20-30 мкл с молярной концентрацией витамина 300 мг/дм3.)
10) Повторить операции последовательно от 4) до 6) три раза.
11) Измерять высоты анодных пиков витамина в пробах с добавкой аттестованных смесей (АС).
12) Вылить содержимое стаканчиков.
13) Записать результаты измерений анализа проб по форме: определяемый компонент (витамин); анализируемая проба (характеристика, номер, дата); условия измерений (чувствительность, объём аликвоты); высота пика анодного тка компонента в пробе, А; добавка; высота пика анодного тока компонента после добавки, А.
14) Рассчитать содержание витамина (мг/кг) в пробе по формуле 3.9.
I1∙ Cд ∙ Vд ∙Vпр
Xi = ------------------ (3.9),
(I1-I2) ∙ m ∙Vал
где: i - номер повторности 1, 2 и т.д.;
I1- величина максимального анодного тока витамина в анализируемой пробе, А;
Cд – концентрация аттестованной смеси АС витамина, из которой делается добавка к анализируемой пробе, мг/дм3;
Vд - объём добавки аттестованной смеси АС витамина, см3;
Vпр – объём разведённой анализируемой пробы, см3;
Vал – объём аликвоты анализируемой пробы, взятой для ВА измерений, см3.
15) Результаты считают статистически достоверными при выполнении условия для двух параллельных определений: [Х1-Х2] ≤ r, мг/кг, или мг/100 г . При превышении предела повторяемости r необходимо дополнительно получить ещё два результата параллельных определений.
16) Представить результаты в виде: (Xcр±∆), мг/кг или мг/100 г продукта, Р=0,95.
3.3 Методы оптического анализа
3.3.1 Фотоколориметрический метод анализа. Закон Бугера-Ламберта-Бера
Свет представляет собой электромагнитное излучение с различными длинами волн или поток фотонов различной энергии. Оптический спектр охватывает ультрафиолетовую, видимую и инфракрасную область. Длины волн ( λ ) обычно выражают в нанометрах (нм) или миллимикронах (ммк), иногда в ангстремах) (Å)
1 нм = 1 ммк = 10‾3 мк = 10‾6 мм = 10‾9 м = 10 Å
1 Å = 10‾7 мм = 10‾10 м.
Ультрафиолетовой области соответствуют электромагнитные колебания с длинами волн 200 – 400 нм, видимой области - 400 – 700 нм, инфракрасной - 0,7 – 100 мк.
Классификация оптических (приборных) методов химического анализа основана на эффекте взаимодействия света с веществом, метод анализа, используемый прибор:
- излучение света - атомно-эмиссионный спектральный анализ (эмиссионная фотометрия пламени), атомно-эмиссионный спектрометр, люминесцентный анализ, флуориметр;
- поглощение света - атомно-абсорбционный анализ, атомно-абсорбционный спектрометр;
- спектрофотометрия в УФ и видимой области спектра, спектрофотометр - фотоэлектроколориметрия, фотоколориметр, ИК-спектроскопия, ИК-спектрометр;
- преломление света - рефрактометрия, рефрактометр, сахариметр;
- рассеяние света - нефелометрия, нефелометр, турбидимегрия, турбидеметр;
- вращение плоскости поляризации - поляриметрический метод анализа, поляриметр.
Вещества в той или иной степени способны поглощать электромагнитные излучения, причем поглощение зависит от природы вещества и его концентрации. Поэтому поглощение света в ультрафиолетовой, видимой и инфракрасной областях спектра имеет большое значение для химического анализа. В частности, в фотоколориметрии широко используется то, что окрашенные растворы поглощают видимую часть спектра.
Абсорбционная спектроскопия основана на избирательном поглощении световой энергии частицами, молекулами или ионами вещества в растворе (рис. 3.2.).
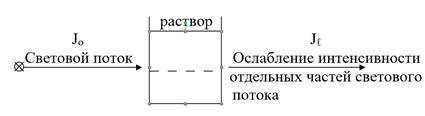
Рис. 3.2. Поглощение световой энергии веществом в растворе
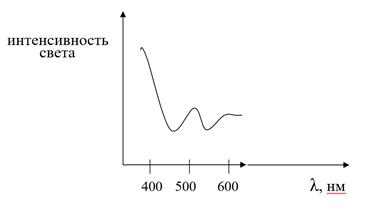
Рис. 3.3. Спектр поглощения