
Лабораторная работа № 8. Определение хлорид-ионов








в почве
Цель работы: определить содержание хлорид-ионов в почвенных образцах различными методами.
Теоретическое обоснование
Настоящий стандарт устанавливает методы определения иона хлорида в водной вытяжке из засоленных почв при проведении почвенного контроля за состоянием солевого режима почв, а также при других изыскательских и исследовательских работах.Сущность метода определения иона хлорида аргентометрическим методом по Мору заключается в титровании иона хлорида в водной вытяжке раствором азотнокислого серебра, образующим с ионом хлорида труднорастворимое соединение. Для установления конечной точки титрования в раствор добавляют хромат калия, образующий с избытком серебра осадок, вызывающий переход окраски раствора от желтой к красно-бурой. Метод не применяют для анализа темно-окрашенных вытяжек. В таких случаях применяют метод прямой ионометрии или метод титровании иона хлорида в водной вытяжке раствором азотнокислого серебра, образующим с ионом хлорида труднорастворимое соединение. Индикацию конечной точки титрования проводят ионометрически с помощью хлоридного ионоселективного электрода (рис. 2.3).
В таких случаях применяют метод прямой ионометрии или метод титровании иона хлорида в водной вытяжке раствором азотнокислого серебра, образующим с ионом хлорида труднорастворимое соединение. Индикацию конечной точки титрования проводят ионометрически с помощью хлоридного ионоселективного электрода (рис. 2.3).
| Рис. 2.3. Установка для ионометрии |
Сущность метода заключается в определении разности потенциалов хлоридного ионоселективного и вспомогательного электродов, значение которой зависит от концентрации иона хлорида в растворе. В качестве вспомогательного электрода используют насыщенный хлорсеребряный электрод. Для предотвращения загрязнения анализируемого раствора хлористым калием из солевого контакта вспомогательного электрода применяют переходную электролитическую ячейку, заполненную раствором азотнокислого калия концентрации 1 моль/дм3. Приборы, реактивы, посуда: аналитические весы; дозаторы; пипетки и бюретки; посуда лабораторная мерная; колбы конические вместимостью 250 см3; 10%-ный раствор К2Сr2О7; 0,1 н раствор КС1; 0,02 моль/дм3 раствора азотнокислого серебра; вода дистиллированная.Подготовка к анализу. Приготовление 0,1 моль/дм3 раствора хлорида калия: 7,456 г хлористого калия, прокаленного до постоянной массы при температуре 500 °С, взвешивают с погрешностью не более 0,001 г, помещают в мерную колбу вместимостью 1000 см3 и растворяют в дистиллированной воде, доводя объем раствора до метки. Приготовленный раствор тщательно перемешивают. Раствор хранят в склянке с притертой пробкой не более 1 года. В случае помутнения, образования хлопьев, осадка, раствор заменяют свежеприготовленным. Для приготовления раствора допускается использовать стандарт-титр хлористого калия или хлористого натрия.Приготовление 0,02 моль/дм3 раствора азотнокислого серебра: 3,4 г азотнокислого серебра, взвешенного с погрешностью не более 0,1 г, помещают в мерную колбу вместимостью 1000 см3 и растворяют в дистиллированной воде, доводя объем до метки. Точную концентрацию раствора проверяют титрованием. Для этого отбирают 10 см3 раствора хлорида концентрации 0,01 моль/дм3 в коническую колбу, приливают 1 см3 раствора хромовокислого калия с массовой долей 10% и титруют раствором азотнокислого серебра до перехода окраски от желтой к красно-бурой. Титрование проводят три раза и для расчета точной концентрации используют среднее арифметическое результатов трех титрований. Точную концентрацию раствора азотнокислого серебра (X), моль/дм3, вычисляют по формуле: 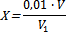 где 0,01 – концентрация раствора хлорида, взятого для титрования, моль/дм3; V – объем раствора хлорида, взятый для титрования, см3; V1 – объем раствора азотнокислого серебра, пошедший на титрование, см3.Раствор хранят в склянке оранжевого стекла с притертой пробкой. Концентрацию раствора проверяют титрованием не реже одного раза в неделю.Ход анализа Для анализа используют фильтраты водных вытяжек. Пробу водной вытяжки объемом от 2 до 20 см3 отбирают дозатором или пипеткой в коническую колбу, приливают дистиллированную воду до объема 20–30 см3, 1 см3 раствора хромовокислого калия с массовой долей10% и титруют раствором азотно- кислого серебра до перехода окраски от желтой к красно-бурой.Объем пробы вытяжки устанавливают по величине удельной электрической проводимости или по величине плотного остатка: 20 см3 – при удельной электрической проводимости вытяжки до 1,5 мСм/см или массовой доле плотного остатка до 0,7%; 10 см3 – при удельной электрической проводимости 1,5–3 мСм/см или массовой доле плотного остатка 0,7–1,5%; 2 см3 – при удельной электрической проводимости свыше 3 мСм/см или массовой доле плотного остатка свыше 1,5%. Обработка результатов Количество эквивалентов иона хлорида (X), ммоль в 100 г почвы,вычисляют по формуле:
где 0,01 – концентрация раствора хлорида, взятого для титрования, моль/дм3; V – объем раствора хлорида, взятый для титрования, см3; V1 – объем раствора азотнокислого серебра, пошедший на титрование, см3.Раствор хранят в склянке оранжевого стекла с притертой пробкой. Концентрацию раствора проверяют титрованием не реже одного раза в неделю.Ход анализа Для анализа используют фильтраты водных вытяжек. Пробу водной вытяжки объемом от 2 до 20 см3 отбирают дозатором или пипеткой в коническую колбу, приливают дистиллированную воду до объема 20–30 см3, 1 см3 раствора хромовокислого калия с массовой долей10% и титруют раствором азотно- кислого серебра до перехода окраски от желтой к красно-бурой.Объем пробы вытяжки устанавливают по величине удельной электрической проводимости или по величине плотного остатка: 20 см3 – при удельной электрической проводимости вытяжки до 1,5 мСм/см или массовой доле плотного остатка до 0,7%; 10 см3 – при удельной электрической проводимости 1,5–3 мСм/см или массовой доле плотного остатка 0,7–1,5%; 2 см3 – при удельной электрической проводимости свыше 3 мСм/см или массовой доле плотного остатка свыше 1,5%. Обработка результатов Количество эквивалентов иона хлорида (X), ммоль в 100 г почвы,вычисляют по формуле: 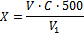 где V – объем раствора азотнокислого серебра, пошедший на титрование, см3; C – концентрация раствора азотнокислого серебра, ммоль/см3; 500 – коэффициент пересчета на 100 г почвы; V1 - объем пробы водной вытяжки, см3.Массовую долю иона хлорида в почве (Х1) в процентах вычисляют по формуле:Х1 = С·0,0355, где С – количество эквивалентов иона хлорида в почве, моль в 100 г; 0,0355 – коэффициент пересчета в проценты.За результат анализа принимают значение единичного определения иона хлорида. Результат выражают в миллимолях в 100 г почвы и в процентах с округлением до трех значащих цифр.
где V – объем раствора азотнокислого серебра, пошедший на титрование, см3; C – концентрация раствора азотнокислого серебра, ммоль/см3; 500 – коэффициент пересчета на 100 г почвы; V1 - объем пробы водной вытяжки, см3.Массовую долю иона хлорида в почве (Х1) в процентах вычисляют по формуле:Х1 = С·0,0355, где С – количество эквивалентов иона хлорида в почве, моль в 100 г; 0,0355 – коэффициент пересчета в проценты.За результат анализа принимают значение единичного определения иона хлорида. Результат выражают в миллимолях в 100 г почвы и в процентах с округлением до трех значащих цифр.
Лабораторная работа № 9. Фотометрический метод
определения ионов марганца в почве
Цель работы: определить фотометрическим методом содержание ионов марганца в образцах почвы.
Теоретическое обоснование
Одной из важнейших групп токсикантов, загрязняющих почву, яв-яются тяжелые металлы (ТМ). К ним относятся металлы с плотностью более 8 тыс. кг/м3 ( кроме благородных и редких ) : РЬ, Сu, Zn, Ni, Cd, Hg, Со, Sb, Sn, Be. В прикладных работах к cписку тяжелых металлов нередко добавляют также Рt, Ag, W, Fe, Mn. Почти все тяжелые металлы токсичны.
В почве протекают различные физические, химические и биологические процессы, которые в результате загрязнения нарушаются. Загрязнение почвы связано с загрязнением атмосферы и гидросферы. В почву попадают твердые и жидкие промышленные, сельскохозяйственные и бытовые отходы. Основными загрязняющими веществами являются металлы и их соединения, удобрения и пестициды, радиоактивные отходы. По пищевым цепям эти загрязнения попадают в организм человека, оказывая токсическое, канцерогенное, мутагенное действие, подавляя иммунитет.
Особую биологическую опасность среди загрязняющих веществ представляют тяжелые металлы (ТМ), к ним относится более 40 химических элементов таблицы Д.И. Менделеева. Здесь следует особо выделить хром, марганец, железо, кобальт, никель, медь, цинк, кадмий, олово, сурьму, теллур, ртуть, галлий, свинец, висмут.
Поступление тяжелых металлов в литосферу вследствие техногенного рассеяния осуществляется разнообразными путями. Важнейшим из них является выброс при высокотемпературных процессах (черная и цветная металлургия, обжиг цементного сырья, сжигание минерального топлива). Кроме того, источником загрязнения биоценозов могут служить орошение водами с повышенным содержанием ТМ, внесение осадков бытовых сточных вод в почвы в качестве удобрения, вторичное загрязнение вследствие выноса ТМ из отвалов рудников или металлургических преприятии водными или воздушными потоками, поступление больших количеств ТМ при постоянном внесении высоких доз орган ческих, минеральных удобрений и пестицидов.
Несмотря на значительное разнообразие соединений ТМ, потупающих в почву из окружающей среды, фазовый состав элементов в составе газопылевых выбросов предприятий цветной металлургии довольно однотипен; они представлены преимущественно оксидами.
Первым этапом трансформации оксидов ТМ в почвах является взаимодействие их с почвенным раствором и его компонентами. даже в такой простой системе, как вода, находящаяся в равновесии с СО2 атмосферного воздуха, оксиды ТМ подвергаются изменениям и существенно различаются по своей устойчивости.
Процесс трансформации тяжелых металлов, поступивших в почву в процессе техногенеза, включает следующие стадии:
1) преобразование оксидов ТМ в гидроксиды (карбонаты, гидрокарбонаты);
2) растворение гидроксидов (карбонатов, гидроксокарбонатов) ТМ и адсорбция соответствующих катионов ТМ твердыми фазами почв;
3) образование фосфатов ТМ и их соединений с органическими веществами почвы.
Часть техногенных выбросов тяжелых металлов, поступающих в атмосферу в виде тонких аэрозолей, переносится на значительное расстояние и вызывает глобальное загрязнение. Другая часть с гидрохимическим стоком попадает в бессточные водоемы, где накапливается в водах и донных отложениях и может стать источником вторичного загрязнения. Основная масса выбросов осаждается в непосредственной близости от источника загрязнения. Теоретически техногенные аномалии представляют систему концентрических колец, в которых концентрация ТМ убывает от центра к периферии. В реальной природной обстановке форма и размеры зон загрязнения существенно отличаются от теоретических; обычно наблюдается неплохая корреляция формы и размеров зон загрязнения с конфигурацией розы ветров. Вокруг крупных предприятий цветной металлургии образуются сильные техногенные аномалии металлов, на пример вокруг Норильского горного металлургического комбината. Для таких предприятий характерно наличие зоны максимальных концентраций тяжелых металлов на расстоянии до 5 км от источника и зоны повышенных содержаний на расстоянии до 20–50 км.
В зонах максимального загрязнения нередко формируется "техногенная пустыня" территория сильно эродированная, личная верхнего гумусового горизонта, растительности. Вокруг промышленных предприятий меньшей мощности зона максимального загрязнения простирается на расстояние до 1–2 км, и площадь загрязненных земель значительно меньше.
Локальные техногенные геохимические аномалии образуются также вокруг предприятий, которые перерабатывают сырье, содержащее тяжелые металлы и другие загрязняющие вещества в виде примесей. Так, геохимические аномалии меди, цинка, свинца образуются вокруг суперфосфатных заводов. Вокруг крупных тепловых электростанций образуются зоны загрязнения металлами 10–20 км в диаметре. Любые городские территории являются значительным источником загрязнения ТМ.
Вблизи автострад обнаружено сильное загрязнение ТМ, особенно свинцом, а также цинком, кадмием. Ширина придорожных аномалий свинца в почве достигает 100 м и более.
Определение степени загрязнения почв тяжелыми металлами не представляется простой задачей. Главная причина заключается в том, что любые элементы в почве присутствуют в форме различных соединений, только часть которых доступна растениям. Но эти соединения могут трансформироваться и переходить из одних форм в другие, поэтому для целей мониторинга выбирают в известной мере условно две или три важнейшие группы. Обычно определяют общее (валовое) содержание элементов или подвижные формы соединений, иногда отдельно определяют обменные формы и водорастворимые соединения.
Химические элементы неравномерно распределены по органам растений. Значительная часть элементов накапливается в наземных частях растений (листьях, стеблях): Mn, Mo, Sr, La, Cu, Ti, Ni, в меньшей степени Fe, Al, Co. В корнях растений аккумулируются такие элементы, KaKAg, Pb, Sn, W, Cr, V, U. Равномерно распределены в органах растений цинк (в растительности таежной зоны), олово и цинк (в альпийских и субальпийских лугах), хром (в растительности альпийских регионов).
Валовое содержание тяжелых металлов определяют методом эмиссионного спектрального анализа без предварительного разложения пробы почвы или методами атомно-абсорбционной спектрометрии после переведения пробы почвы в раствор путем разложения кислотами.
В случае эмиссионного спектрального анализа навеску почвы в 5–10 г растирают в агатовой ступке до состояния пудры. Из растертой пробы берут навеску около прокаливают ее в муфельной печи при 450–500°С в течение 2 ч для удаления воды и разложения органического вещества. Определяют потерю от прокаливания для пересчета результатов анализа на исходную массу почвы. В ходе анализа пробу почвы сжигают в дуге переменного тока, и эмиссионные спектры регистрируют на фотопленке. Для сжигания пробы почвы ее набивают в канал (концентрическую выточку) нижнего электрода спектрографа, верхний электрод изготовлен в форме усеченного конуса с площадкой на торце диаметром около 1 мм. Электроды устанавливают в держатели спектрографа, начинают сжигание при небольшой силе тока в 3–5 А, а затем повышает ее до 18–20 А и проводят сжигание в течение 2,5–3 мин до полного испарения пробы.
Для определения подвижных соединений тяжелых металлов используют кислотные, солевые и водные вытяжки из почв.
Соединения марганца являются сильными окислителями. Предельно допустимая концентрация 2 мг/кг почвы. Для определения содержания тяжелых металлов в почве чаше используют атомно-абсорбционный и полярографический методы. В данном случае используется фотометрический метод определения, который основан на окислении ионов марганца персульфатом аммония в сернокислом растворе.
Приборы, реактивы, посуда: фотоэлектрокалориметр (λ=530 нм), сито с диаметром пор 1 мм, кюветы с толщиной слоя 50 мм, аппарат для встряхивания, ступка фарфopовая, пипетки, колбы конические, стаканы химические, воронки стеклянные, фильтры бумажные «синяя лента», растворы серной кислоты 10%-ный, 5%-ный и 0,1 н., азотная кислота (ρ = 1,4 г/см3), 30%-ный раствор пероксида водорода, ортофосфорная кислота (ρ = 1,7 г/см3) 1%-ный раствор нитрата серебра, персульфат аммония, растворы перманганата калия 0,1 н., 0,001 н. (1мл 0,001 н. раствора содержит 11 мг Мn).
Ход анализа
1. Почву доводят до воздушно-сухого состояния, просеивают через сито с диаметром пор 1 мм. Помещают 5 г почвы в колбу с притертой пробкой, приливают 50 мл 0,1 н. раствора серной кислоты и встряхивают в течение 1 ч.
2. Смесь фильтруют, 10 мл фильтрата помещают в стакан вместимостью 50 мл, приливаю 5 мл азотной кислоты и 2 мл пероксида водорода, нагревают до образования сухого остатка. Затем остаток растворяют в .5 мл 10%-ного раствора серной кислоты, нагревая до полного растворения. К раствору приливают 15 мл воды, 2 мл раствора нитрата серебра и 2 мл фосфорной кислоты. Смесь нагревают 5–10 мин на электрической плитке. Если раствор помутнеет, отфильтровывают. Затем прибавляют 2 г персульфата аммония, перемешивают и ставят на горячую плитку на 10–15 мин. По окончании выделения пузырьков озона раствор охлаждают, переливают в мерную колбу на 50 мл, доводят объем дисводой до метки.
3. Измеряют оптическую плотность раствора при λ = 530 нм по отношению к 5%-ному раствору серной кислоты. Содержание перманганата калия (мл) при анализе пробы находят по градуировочному графику.
4. Построение градуировочного графика.
В мерные колбы вместимостью 50 мл вносят 2,0; 5,0; 10,0; 20,0; 25,0 мл 0,001 н. раствора перманганата калия и объем доводят до метки водой. Измеряют оптическую плотность окрашенных растворов при λ=530 нм. Строится градуировочный график зависимости оптической плотности от объема 0,001 н. перманганата калия, мл.
Обработка результатов
Концентрация марганца в пробе С, мг/кг, вычисляют по формуле
С= а·11/ В ,
где а – содержание 0,001 н. раствора перманганата калия, найденное по градуировочному графику; В – масса почвы, соответствующая исследуемому объему раствора пробы ; 11 – содержание марганца в 1 мл 0,001н. раствора перманганата калия, мкг/мл.
Лабораторная работа № 10. Определение
поверхностно-активных веществ ( ПАВ ) в почвах
Цель работы: определить содержание анионоактивных ПАВ в образцах почвы.
Теоретическое обоснование
В настоящее время насчитывается около 4000 областей применения ПАВ. Недостатком ПАВ является невозможность извлечения из сточных вод для использования в технологическом процессе. Попадая в водоемы, а затем и в почву они могут отрицательно влиять на биологические и биохимические процессы в них.
Основная нагрузка в процессе самоочищения почвы от загрязнителей ложится на микроорганизмы. В этой связи обращает внимание способность ПАВ изменять количественный и качественный состав микрофлоры почвы. Имеются данные об ингибирующем действии алкилбензосульфонатов на процесс нитрификации, проявление которого в различных почвах зависит от степени разветвленности алкильной цепи ПАВ. Анионное ПАВ алкилсульфонат натрия угнетает целлюлозо-разлагающую активность микроорганизмов. Имеется ряд других примеров отрицательного влияния детергентов на микрофлору почвы. ПАВ внедряются в пищевые цепочки, загрязняют продовольственное сырье и продукты питания, оказывая неблагоприятное воздействие на здоровье человека. ПАВ способны образовывать в почве нитрозосоединения. В сточных водах, предназначенных для орошения, обнаружено и идентифицировано около 200 ПАВ.
Методика определения предназначена для суммарного содержания анионоактивных ПАВ в почве, куда входят детекторы естественного и экзогенного происхождения : соли карбоновых кислот СООН, алкилосульфонаты первичные ROSO3Na, вторичные R2CHOSO3Na, сульфонаты карбоновых кислот RCH(S03Na)COONa, их эфиры и амиды, алкилсульфонаты фосфор- и кремнийорганических соединений анионного типа.
Предельно допустимая концентрация ПАВ в почвах составляет 0,2 мг/кг почвы.
Приборы, посуда, реактивы: фотоэлектроколориметр с λ= 625–750 нм, кювета с толщиной слоя 3 см, сушильный шкаф, делительные воронки; ступка фарфоровая, пипетки, раствор гидроксида натрия (5,04 г NaОН в 630 мл воды ), раствор KH2PO4 (16,3306 г KH2PO4 в 1,2 л воды), фосфатный буферный раствор с рН = 10 ( готовят смешением равных количеств растворов NaОН и KH2PO4 ), метиленовый синий ( водный раствор 0,35 г/л), серная кислота ( р = 1,84 г/см3), метиленовый синий (0,35 г МС в 500 мл воды в мерной колбе на 1 л, добавляют 6,5 мл концентрированной сepой кислоты, до метки доводя водой), хлороформ, этанол 70%-ный водный раствор, алкилсульфонат натрия (0,5 г ПАВ в мерной колбе на 1 л).
Ход анализа
1. Методом квартования отбирают часть средней пробы (100–150 г)и высушивают в сушильном шкафу при температуре 105°С до постоянной массы (3–4 ч ). Затем почву растирают до состояния пудры в фарфоровой ступке, навеску массой 1 г переносят в коническую колбу вместимостью 100 мл. В колбу приливают 25 мл 70%-ного раствора этанола нагретого до кипения, и смесь перемешивают в течение 3 мин. Экстракт фильтруют в мерную колбу вместимостью 100 мл, объем фильтрата доводят до метки дистиллированной водой и перемешивают.
2. Раствор переносят в делительную воронку вместимостью 250 мл, добавляют 10 мл фосфатного буферного раствора, 5 мл водного раствора метиленового синего, перемешивают и оставляют на 15 мин. Затем до-бавляют 8 мл хлороформа в воронку и интенсивно встряхивают 1 мин. После расслоения жидкостей хлороформный слой переносят в другую делительную воронку, содержащую 110 мл дисводы и 5 мл кислотного метиленового синего. Смесь встряхивают 1 мин и оставляют для расслоения жидкостей. Хлороформный слой фильтруют через воронку с плотным фильтром в пробирку с притертой пробкой. Извлечение ПАВ из водного раствора повторяют. Хлороформное экстракты объединяют и измеряют оптическую плотность окрашенного раствора на фотоэлектрокалориметре при λ= 625–750 нм. Содержание ПАВ находят по калибровочнону графику.
3. Построение калибровочного графика.
В мерные колбы вместимостью 100 мл вносят 0; 2; 5; 10; 15; 20; 25 мл рабочего стандартного раствора ПАВ, объем доводят до метки водой. Измеряют оптическую плотность окрашенных растворов-экстрактов по отношению к контрольной пробе, не содержащей ПАВ, по средним результатам строят график зависимости оптической плотности от содержания ПАВ (мкг/кг).
Обработка результатов
Концентрацию анионоактивных ПАВ С, мг/кг, вычисляют по формуле
С = а/в,
где а – количество исследуемого ПАВ, найденного в пробе, мкг; в – масса исследуемой почвы, г.