
Вычисление рН при гидролизе солей








Гидролиз солей представляет собой реакцию взаимодействия ионов соли с водой, в результате которой образуются слабые электролиты. Раствор нейтрального соединения – соли – приобретает при этом либо кислую, либо щелочную реакцию. Как известно, соли в свою очередь образуются в результате реакций нейтрализации, при взаимодействии кислот и оснований. Из всех типов солей гидролизу могут подвергаться только три типа, образующиеся при взаимодействии:
1) слабой кислоты и сильного основания;
2) сильной кислоты и слабого основания;
3) слабой кислоты и слабого основания.
Четвертый тип солей, образующихся при взаимодействии сильного основания и сильной кислоты, например NaOH и HCI, по реакции
NaOH + HCI = NaCl + Н2О
гидролизу не подвергаются, так как образующаяся соль NaCl также является сильным электролитом и в водном растворе её молекулы нацело диссоциируют на гидратированные (т.е. окруженные молекулами воды) ионы Na+ и Сl-. При этом равновесие 2Н2О ↔ НЗО+ + ОН- не нарушается и, следовательно, гидролиза не происходит, раствор остается нейтральным. Величина рН такого раствора равна 7.
Рассмотрим примеры гидролиза каждого типа солей в отдельности.
1. Если соль образована слабой уксусной кислотой CH3COOН и сильным основанием NaОН, например ацетат натрия CH3COONa, то уравнение гидролиза запишется так:
● в молекулярной форме
СНЗСООNa + Н2О 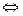 СН3ССОН + NaОНl; (2.8 а)
СН3ССОН + NaОНl; (2.8 а)
● в ионной форме
СНЗСОО-+ Na+ + Н20 CH3COOH+Na+ + ОН-; (2.8 б)
● в сокращенной ионной форме
СНЗСОО- + Н20 CH3COOH + + ОН-. (2.8 в)
Как видно из приведенных уравнений, при гидролизе CH3COONa из-за связывания ацетат-ионами ионов водорода воды в слабую уксусную кислоту в растворе накапливаются ионы ОН-, и рН раствора будет больше 7.
Константа равновесия реакции (2.8.в) запишется в виде:
|
|
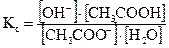 . (2.9)
. (2.9)
Принимая концентрацию воды [H2O] величиной постоянной и объединив её с константой Кс, получим выражение для константы гидролиза:
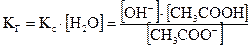 . (2.10)
. (2.10)
Выразив [OH-] через ионное произведение воды 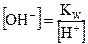 , имеем
, имеем
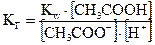 . (2.11)
. (2.11)
Так как в последнем выражении отношение 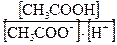
представляет собой величину, обратную константе диссоциации уксусной кислоты 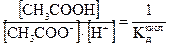 ,
,
выражение для константы гидролиза соли, образованной слабой кислотой и сильным основанием (2.10), запишется следующим образом:
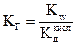 . (2.12)
. (2.12)
Как видно из последней формулы, чем слабее кислота, т.е. чем меньше её константа диссоциации, тем в большей степени соль подвержена гидролизу.
Количественно процесс гидролиза можно охарактеризовать также степенью гидролиза «h», которая представляет собой отношение числа молекул соли, подвергшихся гидролизу, к первоначальному числу молекул. Концентрация той части соли, которая подвергалась гидролизу численно будет равняться концентрации ионов ОН- в растворе, которая, в свою очередь, в соответствии с уравнением (2.8в) равняется концентрации образующейся кислоты, т.е.
[СН3СООН] = [ОН-] = h∙С ,
где С – первоначальная концентрация СН3СООNa, г-моль/л. Концентрация ацетат-ионов [СН3СОО-] будет равна разности
[СН3СОО-] = С - h∙С = С∙(1- h).
С учетом введенной величины h получаем выражение, связывающее константу и степень гидролиза:
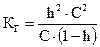 . (2.13)
. (2.13)
При 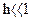 величиной h в знаменателе последнего выражения можно пренебречь, и тогда формула (2.13) запишется следующим образом:
величиной h в знаменателе последнего выражения можно пренебречь, и тогда формула (2.13) запишется следующим образом:
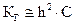 , (2.14)
, (2.14)
откуда 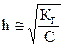 . (2.15)
. (2.15)
Степень гидролиза тем выше, чем более разбавлен раствор, а также чем выше температура, так как с ростом температуры растет KW. Добавление же в раствор ионов ОН-, согласно принципу смещения равновесия Ле-Шателье, будет подавлять процесс гидролиза.
Если соль образована многоосновной кислотой, то гидролиз будет протекать преимущественно по первой ступени. Так, например, уравнение гидролиза соды Na2CO3 запишется следующим образом:
CO32- + H2O ↔ HCO3- + OH-
и константа гидролиза будет определяться величиной константы диссоциации угольной кислоты по первой ступени:
Н2СО3 ↔ H+ + HCO3-
Для получения формулы для расчета рН растворов, образующихся в результате гидролиза, пребразуем выражение (2.10), для чего примем, что величина концентрации ацетат-ионов из-за очень малой степени гидролиза практически равна исходной концентрации соли С. Тогда получим 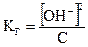 , (2.16)
, (2.16)
т.е. концентрация ионов гидроксила [ОН-], образовавшихся в результате гидролиза, равна [ОН-] = С. (2.17)
Если воспользоваться в данном случае оператором р≡ -lg, то это выражение запишется в виде
pОН =-lg 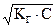 =
= 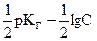 , (2.18)
, (2.18)
или, учитывая выражения (2.7. и 2.12)
рН = 14 - 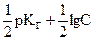 = 7 +
= 7 + 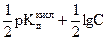 . (2.19 )
. (2.19 )
2. Если соль образована сильной кислотой и слабым основанием,
NH40H + HCl, = NH4Cl + Н2О,
то уравнение гидролиза запишется следующим образом:
● в молекулярной форме
NН4Сl + Н20 = NН40Н + HCl; (2.20 а)
● в ионной форме
NH+4 + Cl-, + 2Н20 = NН40Н + H30+ + Cl- ; (2.20 б)
● в сокращенной ионной форме
NH+4 + 2Н20 = NН40Н + H30+ . (2.20 в)
Константа гидролиза в этом случае имеет вид
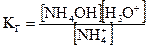 . (2.21)
. (2.21)
Если умножить числитель и знаменатель этого уравнения на [ОН-], то выражение для КГ примет вид
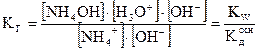 . (2.22)
. (2.22)
В случае разбавленного раствора можно принять, что концентрация гидролизованной части соли, считаемая по [Н30+], равна концентрации основания, т.е. [H3O+] = [NH4OH], а концентрация ионов [NH4+] равна концентрации соли (С). Тогда 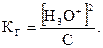 (2.23)
(2.23)
Следовательно, концентрация ионов гидроксония, образовавшихся при гидролизе, равна
[H3O+] = 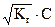 . (2.24)
. (2.24)
Воспользовавшись величиной р = - lg; получим
pH = 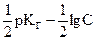 = 7 -
= 7 - 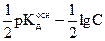 . (2.25)
. (2.25)
Степень гидролиза
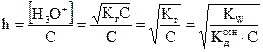 . (2.26)
. (2.26)
Таким образом, чем слабее основание (чем меньше 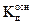 ), тем больше концентрация ионов [H30+] в растворе, т.е. тем сильнее протекает гидролиз соли, образованной сильной кислотой и слабым основанием. Добавляя в раствор ионы [H30+], можно ослабить или совсем предотвратить процесс гидролиза, так как согласно уравнению (2.20 в) равновесие при этом смещается влево.
), тем больше концентрация ионов [H30+] в растворе, т.е. тем сильнее протекает гидролиз соли, образованной сильной кислотой и слабым основанием. Добавляя в раствор ионы [H30+], можно ослабить или совсем предотвратить процесс гидролиза, так как согласно уравнению (2.20 в) равновесие при этом смещается влево.
3. Гидролиз соли, образованной слабым основанием и слабой кислотой, например ацетата аммония СНЗСООNН4 по схеме
CH3COONH4+H2O ↔ CH3COOH+NH4OH,
протекает практически полностью.
Константа гидролиза
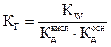 . (2.27)
. (2.27)
pH такого раствора зависит только от величин констант диссоциации кислоты и основания и не зависит от концентрации соли:
[H3O+]= 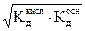 (2.28)
(2.28)
и 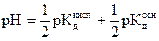 . (2.29)
. (2.29)
Таким образом, соли подвергаются гидролизу, если в результате образуется более слабый электролит, чем исходное соединение.
2.4. Буферные растворы
Буферными называются водные растворы электролитов, которые сохраняют практически неизменным значение рН при разбавлении или добавлении небольших количеств кислоты или щелочи. Буферные растворы представляют собой смесь либо слабой кислоты и соли, образованной этой кислотой и сильным основанием, либо слабое основание и соль, образованную этим основанием и сильной кислотой.
Если, например. добавить к раствору слабой уксусной кислоты СН3СООН соль, которая содержит тот же анион (например, ацетат натрия СНЗСООNa), то, согласно принципу Ле-Шателье, равновесие процесса диссоциации кислоты
CH3СOOH ↔ СН3СОО- + Н+ (2.30)
будет сдвинуто влево, что практически полностью подавит процесс диссоциации кислоты и степень диссоциации α будет равна нулю (α = 0).
Соль же будет диссоциирована нацело согласно уравнению
СНЗСООNa ↔ СН3СОО- + Na+ (2.31)
В этом случае в смеси кислоты и соли концентрация недиссоциированных молекул кислоты будет равна исходной концентрации кислоты Скисл, а концентрация ацетат-ионов СН3СОО- – исходной концентрации соли Ссоли .
Если поставить эти величины в выражение для константы диссоциации кислоты
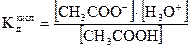 , (2.32)
, (2.32)
то концентрация ионов [НЗО+] в растворе будет равна
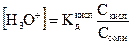 (2.33)
(2.33)
и рН такого раствора можно рассчитать по формуле
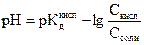 . (2.34)
. (2.34)
Таким образом, для того чтобы определить рН буферного раствора, составленного из слабой кислоты и соли, образованной этой кислотой и сильным основанием, надо знать только первоначальные кон-
центрации этих компонентов.
Для смеси слабого основания NH4OH и его соли NH4Cl, анион которой является анионом сильной соляной кислоты, воспользовавшись предыдущими рассуждениями, можно показать, что кислотность такого раствора будет выражаться уравнением
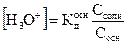 , (2.35)
, (2.35)
а рН буферной смеси – уравнением
pH=p 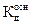 - lg
- lg 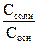 . (2.36)
. (2.36)
На основании вышеприведенных зависимостей видно, что рН буферных растворов не зависит от разбавления, так как в этом случае в равной степени меняются и концентрация кислоты, и концентрация соли (или основания и соли), при этом их отношение остается неизменным. Это первая отличительная особенность буферных растворов.
Если добавить к буферному раствору небольшие количества кислоты или щелочи, то рН этих растворов будет изменяться очень незначительно. Это их вторая отличительная особенность.
Например, если к ацетатному буферному раствору, содержащему смесь СНЗСООН и CH3COONa, добавить небольшое количество HCI, то ацетат натрия будет взаимодействовать с соляной кислотой, полностью диссоциирующей в растворе на ионы Н3О+ и Cl- по схеме
CH3COO- + Na+ + H3O- + Cl- ↔ CH3COOH + Na+ + Cl-. + Н2О
Изменение концентрации ионов [Н30+], а следовательно и рН раствора, согласно уравнению (2.36), практически не происходит. Чем меньше изменение рН при добавлении кислоты или основания, тем сильнее буферное свойство раствора. Та область концентраций, в которых рН буферных растворов остается практически неизменной, называется буферной емкостью:
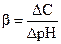 (2. 37)
(2. 37)
Таким образом, буферная емкость представляет собой количество г-эквивалентов кислоты или щелочи, которое можно прилить к 1 л буферного раствора, чтобы изменить значение его рН на единицу. Буферные растворы широко используются для создания стандартных растворов с определенным значением рН при калибровке различных приборов, измеряющих кислотность растворов, например рН-метров.
2.5. Ионообменная хроматография
Хроматография представляет собой метод разделения и анализа смесей, который основан на различном распределении ее компонентов между неподвижной и подвижной фазами. Когда смесь веществ проходит через хроматографическую колонку, заполненную сорбентом (это мелкораздробленная неподвижная фаза), в ней происходят динамические про
цессы сорбции и десорбции молекул подвижной фазы. При этом в разной степени адсорбирующиеся компоненты поглощаются на разной высоте неподвижной фазы, т.е. на разных уровнях хроматографической колонки. Добавив растворитель, можно выделить (вымыть) фракции, содержащие отдельные вещества, ранее входящие в смесь, и таким образом разделить последнюю на компоненты. Наименее сорбируемый компонент смеси (скорость движения которого наибольшая) будет концентрироваться в нижней части хроматографической колонки и первым вымывается растворителем из нее. Хроматографические методы используются для разделения газов, паров и жидкостей в динамических условиях. Существуют различные виды хроматографии: адсорбционная, ионообменная, распределительная и др. Адсорбционная хроматография основана на различной способности компонентов смеси к адсорбции за счет действия межмолекулярных сил. Ионообменная хроматография основана на способности смеси к ионному обмену с сорбентом, происходящему в результате химического взаимодействия молекул (хемосорбция).
В последнем случае сорбент представляет собой высокомолекулярные органические вещества (синтетические смолы), имеющие группы, содержащие либо подвижный ион водорода, либо подвижный ион гидроксила. В первом случае это могут быть сульфогруппы – S03Н, карбоксильные группы – СООН и др., Во втором – анионы ОН-, амминные группы NH2-, NH2- и др. Такие сорбенты называются ионитами. Иониты, обменивающиеся с раствором катионами, называются катионитами, а обменивающиеся анионами — анионитами.
В общем виде реакции химического взаимодействия между ионитами и раствором можно представить следующим образом:
● на катионите
m∙R-H + Mem+ ↔Rm-Me + m∙H+; (2.38)
● на анионите
n∙R-OH + An-↔Rn-A + n∙OH- . (2.39)
Обмен катионов и анионов между ионитами и раствором происходит в количествах, равных или пропорциональных химическим эквивалентам. Химическая реакция обмена ионов является обратимым процессом.
Метод ионообменной хроматографии применяется, в частности, для очистки воды от различных примесей, причем о количестве последних, поглощенных сорбентом, можно судить по изменению электропроводности раствора. Так, при прохождении жидкости через катионит за счет ионного обмена в растворе появляются наиболее подвижные катионы – ионы водорода, вследствие чего удельная электропроводность раствора увеличивается. При пропускании раствора через анионит освобождающиеся ионы ОН- взаимодействуют с ионами водорода и образуют малодиссоциируемое соединение – воду Н20.
Электропроводность раствора при этом уменьшается. Степень очистки раствора можно контролировать и по изменению его кислотности (рН). Вода, очищенная от положительно и отрицательно заряженных ионов методом ионообменной адсорбции, называется деионизированной.
2.6. Коллоидные растворы
Коллоидными растворами называются мелкодисперсные системы, в которых существует граница раздела фаз между растворителем и растворенным веществом, частицы которого независимо друг от друга перемещаются путем броуновского движения. Радиус частиц, входящих в состав таких растворов, находится в пределах 10-7 м > r >10-9 м. Таким образом, коллоидные частицы нельзя увидеть в оптическом микроскопе, разрешающая способность которого ниже этих пределов.
Коллоидные системы относятся к гетерогенным системам, которые содержат две фазы, например твердую и жидкую, равномерно распределенные друг в друге. Они широко распространены в природе, используются во многих производствах. К коллоидным растворам принадлежат многие естественные продукты, такие как молоко, кровь, яичный белок – это жидкие коллоидные системы. Твердые коллоидные системы представляют собой минералы (опалы, яшмы, агаты и др.). Атмосферный туман, вулканический дым – газообразные коллоидные системы.
Жидкие коллоидные системы называются золями. Они делятся на лиофобные (гидрофобные) и лиофильные (гидрофильные). Коллоидные растворы, в которых практически отсутствует взаимодействие между молекулами растворенного вещества и растворителя, называются лиофобными. Примером таких растворов служат коллоидные растворы металлов, раствор серы в воде. Дисперсные системы, для которых характерно интенсивное взаимодействие дисперсной среды с поверхностью дисперсной фазы, называются лиофильными системами. Частицы растворенного вещества (дисперсной фазы) при этом окружены молекулами растворителя (дисперсионной среды), образующими сольватную или гидратную оболочку. Лиофильные системы образуются самопроизвольно, например при диспергировании мыла, глины, полимеров в растворителе под действием теплового движения.
Смачиваемость поверхности какого-либо вещества возможна лишь при использовании растворов, обладающих лиофильными свойствами по отношению к природе поверхности данного вещества. Учитывая тот факт, что протекание любого гетерогенного процесса (травления, осаждения покрытий и др.) начинается с процесса смачивания, очевидна необходимость тщательного подбора реагирующих веществ для подготовки поверхности.
По Н.Д. Пескову, следует различать два вида устойчивости коллоидных систем: кинетическую (седиментационную) и агрегативную.
Кинетическая устойчивость характеризует устойчивость к оседанию частиц при их тепловом (броуновском) движении в растворе. Чем больше масса частиц, тем меньше их кинетическая устойчивость.
Агрегативная устойчивость характеризует устойчивость системы против слипания частиц при их столкновении друг с другом. Она зависит от степени сольватации (гидратации) растворенных частиц, от величины энтропии системы, характеризующей степень ее хаотичности, и от кинетической энергии частиц. Если система теряет агрегативную устойчивость, то коллоидный раствор коагулирует, т.е. происходит слипание частиц с образованием более крупных агрегатов и их осаждение или всплытие на поверхность. При этом происходит и потеря кинетической устойчивости системы.
Элементарная частица коллоидных систем называется мицеллой. Она состоит из ядра, нерастворимого в дисперсионной среде, адсорбционного и диффузного слоев. Ядро представляет собой агрегат многих молекул, которые могут иметь различный состав, например
[Fе(ОН)з]n, [AgJ] n и т.д., где n – число молекул, входящих в состав агрегата, и адсорбционного слоя, который состоит из ионов, преимущественно общих с ионами, составляющими ядро (правило Пескова – Фаянса).
Ядро коллоидной частицы {[Fе(ОН)з]n ∙m Fe+3}+3m заряжено положительно, благодаря адсорбции потенциалопределяющих ионов Fe+3 на поверхности ядра. Так как в целом мицелла электронейтральна, этот положительный заряд уравновешивается равным отрицательным зарядом противоионов, например ионов С1-. Силы взаимодействия в этом случае имеют электростатическую и адсорбционную природу. Те ионы С1-, которые прочно связаны с ядром и входят в состав адсорбционной зоны, называются противоионами адсорбционного слоя и составляют вместе с ядром гранулу (частицу). Те же ионы, которые из-за собственного теплового движения распределяются диффузно, называются противоионами диффузного слоя. Разделение окружающих ядро противоионов на две части – одну, находящуюся внутри границы скольжения и движущуюся совместно с ним, и вторую, «отрывающуюся» под действием внешнего поля от поверхности частицы, – позволяет записывать своебразные «химические формулы», отражающие строение мицелл коллоидных систем. Для гидрозоля гидроксида железа строение мицеллы имеет следующий вид:
{[Fе(ОН)з]n∙mFe+3 ∙ 3(m-x)Cl-}3x+ · 3xCl- (2.40)
ядро адсорбционный диффузный
слой слой
[ ____________________гранула________ ]
[___________________ мицелла _______________]
|
|
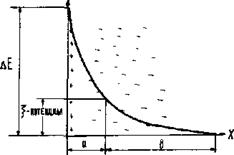
Рис. 2.1. Изменение потенциала в двойном электрическом слое
Из структуры мицеллы видно, что на межфазной границе возникает двойной электрический слой, который обладает определенным потенциалом. Внешний электрический слой состоит из двух частей. Первую составляют противоионы, которые находятся вблизи ядра и удерживаются около него силами электростатического притяжения (толщину этого слоя обозначим буквой «а») (рис. 2.1); вторую часть составляют противоионы, диффузно распределенные по объему (слой «в»).
В слое «а» изменение потенциала носит линейный характер (по аналогии с конденсатором), а в слое «в» (диффузном) – экспоненциальный.
Тот потенциал, который возникает на границе скольжения фаз относительно друг друга (между гранулой и диффузным слоем), называется электрокинетическим (или ξ- потенциалом).
Чем больше концентрация электролита и заряд его ионов, тем меньше толщина слоя «а» и величина ξ -потенциала. Это обусловлено тем, что с ростом числа ионов, определяющих заряд ядра, большее число противоионов переходит из диффузной части двойного электрического слоя в адсорбционную.
Если приложить к коллоидному раствору внешнюю разность потенциалов, то в них наблюдается либо направленное перемещение частиц к электродам согласно знакам их зарядов (явление электрофореза), либо направленное перемещение одной жидкой фазы относительно другой (явление электроосмоса).
Устойчивость коллоидных систем может быть нарушена путем добавления к ним сильных электролитов. Коагулирующее действие электролитов на коллоидные системы характеризуется порогом коагуляции, т.е. наименьшей концентрацией электролита, вызывающей коагуляцию.
При добавлении электролита к лиофобному коллоидному раствору за счет вытеснения противоионов из диффузной части мицеллы в адсорбционную толщина двойного электрического слоя уменьшается. В случае лиофильных коллоидных растворов введение электролита способствует разрушению сольватной оболочки. Силы притяжения между частицами начинают возрастать, и наступает быстрая коагуляция.
Высокомолекулярные соединения (ВМС) и лиофильные коллоиды являются стабилизаторами по отношению к лиофобным золям. Так, если прибавить к коллоидному раствору соли серебра, например AgI, небольшое количество желатина или белка, и восстановить серебро до образования золя, то степень дисперсности коллоидного серебра в этих условиях его получения оказывается более высокой и золь менее подвержен влияниям факторов, вызывающих коагуляцию. Вследствие защитного действия, которое в подобных случаях оказывают лиофильные коллоиды, повышая стабильность золей, их называют защитными коллоидами. То минимальное количество высокомолекулярного вещества, которое необходимо для защиты 10 мл коллоидного раствора от коагуляции, называется защитным числом:
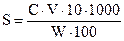 , (2.41)
, (2.41)
где С – процентное содержание защищающего вещества; V – объем защищающего вещества; W – объем коллоидного раствора.
2.7. Электропроводность растворов электролитов
При отсутствии внешнего электрического поля ионы в растворах электролитов беспорядочно перемещаются по всем направлениям. При подключении внешнего источника тока начинает преобладать одно из направлений перемещения ионов согласно знакам из зарядов. В общем случае электропроводность W — величина, обратная электрическому сопротивлению R , в системе СИ имеет размерность Ом-1.
Для измерения электропроводности различных растворов необходимо иметь электрохимическую ячейку, которая состоит из двух проводников первого рода, погруженных в раствор электролита (проводник второго рода) и соединенных с внешним источником тока.
Удельная электропроводность раствора определяется как величина, обратная удельному электрическому сопротивлению 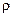 :
:
æ = 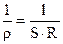 , (2.42)
, (2.42)
где æ — удельная электропроводность, Ом-1, м-1 ; R — сопротивление, Oм; S — площадь поперечного сечения проводника, м2 ; l – длина проводника, м.
Таким образом, удельная электропроводность æ представляет собой количество электричества, которое проходит через поперечное сечение электролита площадью 1 см2 зa 1 c при приложенном внешнем напряжении
1 B и расстоянии между электродами 1 см, т.е. это – электропроводность одного кубического сантиметра электролита. Скорость движения иона в электрическом поле определяется как величина пути его направленного перемещения к одному из электродов и выражается в системе СИ в м/с.
Зависимость удельной электропроводности от концентрации раствора электролита показана на рис. 2.2. Эта зависимость представлена кривыми с максимумом. Такой ход кривых легко объяснить, исходя из следующих рассуждений. На начальном участке кривой с ростом концентрации раствора удельная электропроводность растворов электролитов возрастает. Это закономерно, так как электрический ток в проводниках второго рода обеспечивается за счет движения ионов, и чем их больше, тем выше электропроводность.
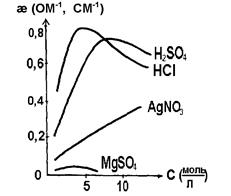
Рис. 2.2. Зависимость удельной электропроводности от концентрации раствора
Однако по мере дальнейшего роста концентрации раствора расстояние между ионами будет уменьшаться и, как следствие, усиливаться взаимодействие между ними, что будет тормозить движение ионов. Поэтому увеличение концентрации раствора после некоторого предела закономерно приведет к уменьшению электропроводности.
С ростом температуры удельная электропроводность возрастает, что объясняется уменьшением вязкости раствора и степени гидратации ионов, а также увеличением степени диссоциации электролита.
В технических расчетах чаще всего пользуются величиной эквивалентной электропроводности. Она обозначается буквой λ и представляет собой количество электричества, протекающее за 1 с по столбику жидкости высотой 1 см и имеющее такое сечение, чтобы в его объеме поместился раствор, содержащий один г-эквивалент растворенного вещества. Падение напряжения на этом столбике должно составлять 1 В.
Для разбавленных растворов эта площадь чрезвычайно велика (например, для 0,01 н раствора она должна быть равной 100 000 см2). Для расчета λ, следовательно, необходимо рассчитать число ячеек площадью 1 см2. Так, для 0,01 н раствора объем, содержащий один эквивалент вещества, будет равен V = 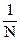 = 100 л (он называется разведением), или 100 000 см3. Поэтому в площади столбика жидкости высотой 1 см уместится 10 000 таких
= 100 л (он называется разведением), или 100 000 см3. Поэтому в площади столбика жидкости высотой 1 см уместится 10 000 таких
ячеек ( ∙ I000), электропроводность каждой из которых равна æ.
Отсюда
λ = (æ/N)∙ I000 см2/г-экв ∙Ом, (2.43)
где N – нормальность раствора.
Обычно на практике измеряют значение удельной электропроводности и по нему рассчитывают эквивалентную электропроводность растворов электролитов.
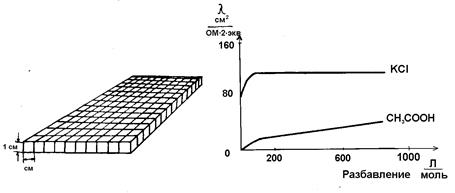
| Рис. 2.3. Графическое изображение взаимосвязи эквивалентной и удельной электропроводности | Рис. 2.4. Зависимость эквивалентной электропроводности от разбавления |
Если использовать величину V, обозначающую разбавление (или – разведение) раствора, то λ = æ∙V∙I000 см2/г-экв∙ Ом. (2.44)
Эквивалентная электропроводность как сильных, так и слабых электролитов увеличивается с ростом разведения и достигает предельной величины, которая называется эквивалентной электропроводностью при бесконечном разведении λ∞ (рис. 2.3). Эта закономерность для растворов слабых электролитов объясняется тем, что процесс диссоциации слабого электролита на ионы с увеличением разбавления может происходить до конца, так как с увеличением разбавления раствора расстояние между частицами растворенного вещества растет и число частиц, распавшихся на ионы, преобладает над количеством недиссоциированных молекул. Поэтому доля распавшихся молекул растворенного вещества в объеме, содержащем его один г-эквивалент, увеличивается. Это приводит к увеличению числа переносчиков электричества, и эквивалентная электропроводность возрастает. Такой рост будет происходить до тех пор, пока все молекулы растворенного вещества не распадутся на ионы (α = I). Строго говоря, это возможно лишь при бесконечном разведении. Такая электропроводность и обозначается λ∞. Следовательно, отношение электропроводностей раствора электролита при данном и бесконечном разведении будет определять степень его диссоциации
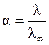 . (2.45)
. (2.45)
Так как электропроводность возникает за счет независимого движения катионов и анионов, то
λ = λ+ + λ- , (2.46)
где λ+ — электропроводность, обусловленная катионами, а λ- — анионами. λ+ и λ- называют также подвижностью катиона и аниона.
Она пропорциональна скорости движения иона V:
λ+ = V+ . F ; ,λ- = V- F,
где F — число Фарадея.
Для бесконечно разбавленного раствора
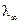 =
= 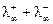 . (2.47)
. (2.47)
Это соотношение выражает собой закон аддитивности электропроводностей Кольрауша. Величины 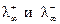 для различных ионов приводятся в справочниках.
для различных ионов приводятся в справочниках.
Возрастание эквивалентной электропроводности с разбавлением раствора (рис. 2.4) для сильных электролитов объясняется тем, что в разбавленных растворах из-за уменьшения межионного взаимодействия, тормозящего движение ионов, облегчается перемещение ионов по всему объему.
Для разбавленных растворов сильных электролитов с концентрацией С < 2∙1O-3 г-экв/л эквивалентная электропроводность зависит от концентрации по уравнению
λ = λ∞ — А 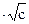 , (2.48)
, (2.48)
где A — постоянный множитель, зависящий от свойств растворителя.
2.8. Кондуктометрическое титрование
Титрование, использующееся в аналитической химии при титриметрическом анализе, в общем случае означает постепенное прибавление определенного количества реагента (например, раствора кислоты) к анализируемому раствору (например, щелочи). Титриметрический анализ представляет собой метод, основанный на измерении объема раствора с известной концентрацией (титрованного раствора), который расходуется на реакцию с данным объемом раствора неизвестной концентрации.
Целью кондуктометрического титрования является определение неизвестной концентрации раствора методом измерения его электропроводности в процессе титрования. При протекании химических реакций происходит обмен ионами, находящимися в растворе, вследствие чего меняется ионный состав раствора и соответственно его удельная электропроводность. Кондуктометрическое титрование чаще всего применяют для исследования реакций, в результате которых образуются нерастворимые или малорастворимые вещества. Этим методом можно исследовать даже мутные и сильноокрашенные растворы.
Общий вид зависимости электропроводности раствора щелочи от количества добавленной кислоты при титровании сильного основания сильной кислотой представлен на рис. 2.5. Точка пересечения двух прямых называется точкой эквивалентности. В этой точке вещества (кислота и щелочь) прореагировали в количествах, пропорциональных их эквивалентам. Для точки эквивалентности справедливо равенство
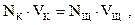 (2.49)
(2.49)
где NK и NЩ — соответственно нормальность растворов кислоты и щелочи раствора); VK и VЩ — объемы растворов кислоты и щелочи, л.
Произведение (N∙V) представляет собой число г-эквивалентов вещества в данном объеме, так как нормальность N – это число г-эквивалентов растворенного вещества в 1 л раствора, а уравнение (2.49) аналитически выражает закон эквивалентов для растворов.
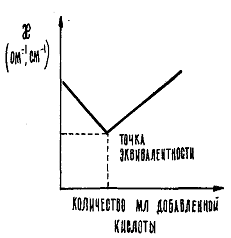
Рис. 2.5. Кривая кондуктометрического титрования
Вид кондуктометрической кривой легко объяснить на примере титрования раствора NaОH раствором HCl, при взаимодействии которых протекает реакция
NaOH + HCl = NaCl + Н2О;
Na+ + ОH- + Н+ + Cl- = Na++ Cl- + Н2О .
При добавлении в раствор щелочи соляной кислоты в ходе реакции происходит образование хлористого натрия NaCl и Н2О. В связи с тем, что в водных растворах молекулы NaOH, HCl, NaCL практически нацело диссоциированы на ионы, в реакционной смеси ионы ОН- будут заменяться на ионы Cl-. Общее число заряженных частиц остается при этом постоянным. Как было показано выше, электропроводность раствора определяется скоростью движения ионов. Если сравнить подвижности указанных ионов, которые при 25oС равны следующим величинам
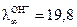 м2/(Ом∙кг-экв); а
м2/(Ом∙кг-экв); а 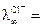 7,63 м2/(Ом∙кг-экв),
7,63 м2/(Ом∙кг-экв),
то становится очевидным, что замена ионов гидроксила на менее подвижные хлорид-ионы должно приводить к уменьшению электропроводности. Вода, являясь малодиссоциирующим соединением, не оказывет практического влияния на изменение электропроводности раствора. В точке эквивалентности (когда все количество щелочи прореагирует и все ионы гидроксила будут связаны в молекулы воды) электропроводность раствора станет минимальной, а затем за счет добавления избытка кислоты она начинает значительно расти. Это связано с тем, что начинает увеличиваться общее количество ионов в растворе, вводимых с избытком кислоты, которая диссоциирует согласно уравнению HCL ↔ Н+ + Cl-, кроме того, подвижность ионов Н+ является самой высокой, при 25oС она равна 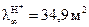 /(Ом∙кг-экв).
/(Ом∙кг-экв).
Кондуктометрический метод используется также для определения растворимости малорастворимых электролитов. Если измерить удельную электропроводность раствора, содержащего малорастворимую соль, то, рассчитав значение эквивалентной электропроводности при бесконечном разведении 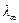 = , можно определить концентрацию раствора по формуле
= , можно определить концентрацию раствора по формуле
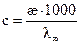 . (2.50)
. (2.50)