
Гетерогенный катализ[править | править вики-текст]








При гетерогенном катализе ускорение процесса обычно происходит на поверхности твердого тела — катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твердый пористый носитель.
Механизм гетерогенного катализа сложнее, чем у гомогенного. Механизм гетерогенного катализа включает пять стадий, причем все они обратимы.
1. Диффузия реагирующих веществ к поверхности твердого вещества
2. Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их
3. Химическая реакция между реагирующими молекулами
4. Десорбция продуктов с поверхности катализатора
5. Диффузия продукта с поверхности катализатора в общий поток
Примером гетерогенного катализа является окисление SO2 в SO3 на катализаторе V2O5 при производстве серной кислоты (контактный метод).
Микрогетерогенный катализ – катализ на макромолекулах или коллоидных частицах (10-5 – 10-7 см), которые имеют огромную удельную поверхность с большим числом активных центров, чем обеспечивается исключительно высокая активность таких катализаторов. Например, коллоидные растворы Pt и Pd проявляют очень высокую активность при пониженных температурах (способствуют энергичному разложению перекиси водорода при концентрации катализатора 10-8 г/л).
Автокатализ — катализ химической реакции одним из её продуктов или исходных веществ. Одним из наиболее широко известных примеров автокатализа является окисление щавелевой кислоты перманганатом:
2MnO4− + 5C2O42− + 16H+ = 2Mn2+ + 10CO2 + 8H2O
Катализатором этой реакции являются ионы Mn2+. При комнатной температуре эта реакция вначале протекает медленно, но по мере накопления в растворе продукта-катализатора, она ускоряется.
Автокатализ играет ключевую роль в эволюционной химии, поскольку реакция, катализируемая собственными продуктами, получает преимущество перед реакциями, получающими катализатор извне (тем более - перед некаталитическими реакциями), что создает условия для естественного отбора.
Вопрос 22
Существенным недостатком теории Аррениуса является и то, чтоона не указывает причин, вызывающих ионизацию электролитов в растворах. Термин «электролитическая диссоциация» подразумевает образование ионов в растворе при распаде нейтральных молекул растворяемого вещества. Однако часто ионы существуют уже до растворения: кристаллы солей построены из ионов, и при растворении должно произойти разрушение кристалла. Расчеты энергии кристаллической решетки DGp (энергия кристаллической решетки – это работа, которую нужно затратить для разрушения решетки, то есть для разведения составляющих ее ионов на бесконечно большое расстояние в вакууме) показывают, что количество термической энергии при обычных температурах слишком мало по сравнению с тем, которое надо затратить на разрушение решетки.
Одно из первых (и одно из наиболее точных) уравнений для подсчета энергии решетки – уравнение Борна (1918):
DGp = NA 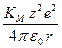
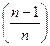 ,
,
КМ – константа Маделунга, зависящая от характера взаимного расположения ионов в кристаллической решетке; ее значения известны для различных типов решетки, например, для NaCl (ГЦК-решетка) КМ = 1,7476;
r – равновесное расстояние между ионами противоположного знака в данном кристалле; обычно оно определяется по принципу плотной упаковки и отвечает сумме кристаллохимических радиусов Гольдшмидта;
n – константа, характеризующая изменение сил отталкивания с расстоянием между частицами; рассчитывается из данных по сжимаемости кристаллов; она лежит в пределах от 5 до 12 (для NaCl n = 7,5):
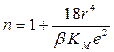 ,
,
где b – коэффициент сжимаемости кристалла.
А.Ф.Капустинский предложил следующие два уравнения (1933, 1943), несколько менее точные, чем формула Борна, но более универсальные и не требующие определения величин КМ и n:
DGp = 1072,2×10–10 n 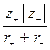 ,
,
DGp = 1202,4×10–10 n – 414,8×10–20 n 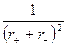 ,
,
где n – число ионов в молекуле данного соединения; r+ , r– – радиусы положительных и отрицательных ионов, м.
Воспользуемся приведенными уравнениями для оценки энергии решетки NaCl, подставив численные значения. Формула Борна дает для энергии решетки
DGp = 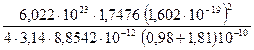
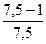 = 762 кДж/моль .
= 762 кДж/моль .
Первая формула Капустинского приводит к величине несколько большей, чем вычисленная по Борну:
DGp = 1072,2×10–10 ×2 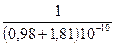 = 769 кДж/моль ,
= 769 кДж/моль ,
а вторая формула Капустинского дает результат несколько меньший:
DGp = 1202,4×10–10 ×2 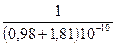 – 414,8×10–20 ×2
– 414,8×10–20 ×2 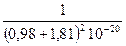 =
=
755 кДж/моль .
Как было показано Борном и Габером (1919), энергию решетки можно найти из термохимических данных, если воспользоваться циклом, основанным на законе Гесса. Подобный цикл можно составить для любого кристаллического вещества. Для NaCl цикл имеет вид:
 NaCl (кр)
NaCl (кр)  Na+(газ) + Cl–(газ)
Na+(газ) + Cl–(газ)
 – е– DGион + е– DGср
– е– DGион + е– DGср
Na (газ) + Cl (газ)
DGобр DGсуб DGдис
Na (тв) + ½ Cl2 (газ)
DGобр – энергия образования хлорида натрия из элементарных натрия и хлора, взятых в их стандартных состояниях (твердый кристаллический натрий и газообразный молекулярный хлор);
DGсуб – энергия сублимации натрия;
DGион – энергия ионизации натрия;
DGдис – энергия диссоциации молекулярного хлора;
DGср – энергия, характеризующая сродство электрона к газообразному атомарному хлору.
DGр = – DGобр + DGсуб + DGион + DGдис + DGср =
– (– 384) + 78 + 496 + 203 – 387 = 774 кДж/моль .
Полученное значение достаточно хорошо согласуется с энергией решетки, подсчитанной по уравнениям Борна и Капустинского.
В кристаллической решетке соседние ионы Na+ и Cl– по сравнению с изолированной молекулой NaCl обладают меньшим числом степеней свободы, поэтому тепловую энергию молекулы NaCl можно рассматривать как ее максимальное значение для пары противоположно заряженных ионов в решетке. Свободная молекула NaCl обладает семью степенями свободы, то есть при 298К запас ее тепловой энергии составляет 8,68 кДж/моль, из которой всего лишь около 2,5 кДж/моль приходится на колебательную энергию, непосредственно вызывающую распад молекул. Это количество термической энергии слишком мало по сравнению с тем, которое надо затратить на разрушение решетки (762 кДж/моль), чтобы обеспечить сколько-нибудь заметную диссоциацию хлорида натрия на ионы. В то же время известно, что в водном растворе NaCl a » 1.
Наиболее полная и четкая формулировка основных недочетов теории Аррениуса и путей их преодоления дана в трудах Д.И.Менделеева. Причина всех недостатков теории заключается в игнорировании взаимодействия частиц растворенного вещества между собой, а также с молекулами растворителя. Менделеев отмечал, что для растворов существенны не только процессы диссоциации, но и процессы образования новых соединений с участием молекул растворителя. Эти взгляды Менделеева были развиты Д.П.Коноваловым, И.А.Каблуковым, В.А.Кистяковским, Л.В.Писаржевским, А.Нойесом и легли в основу современной теории растворов.
В теории Аррениуса ионы рассматривались как частицы идеального газа, а следовательно, не учитывались обусловленные кулоновскими силами притяжение катионов и анионов и отталкивание одноименно заряженных ионов.Пренебрежение ион-ионным взаимодействием, совершенно непонятное с физической точки зрения, и приводило к нарушению количественных соотношений теории Аррениуса, о чем уже говорилось выше (константа диссоциации не оставалась постоянной, а изменялась при изменении концентрации электролита, что особенно отчетливо проявлялось в растворах сильных электролитов – так называемая «аномалия сильных электролитов»; различные методы определения степени диссоциации a давали несовпадающие результаты, а в концентрированных растворах сильных электролитов иногда получались не имеющие физического смысла значения a > 1 и др.).
Если в раствор ввести большой избыток постороннего электролита (так называемого «фона»), который не участвует непосредственно в ионных равновесиях, то в этих условиях основные соотношения теории Аррениуса выполняются с очень хорошей степенью приближения. Метод введения избытка индифферентного электролита был предложен Я.Брёнстедом и получил название метода постоянной ионной среды.
Если игнорировать ион-дипольное взаимодействие (то есть взаимодействие ионов с диполями воды или другого растворителя), то нельзя объяснить процесс образования ионов и устойчивость ионных систем, ведь именно это взаимодействие, как показано далее, является физической основой образования ионов в растворе при растворении электролита. Вопрос о причинах электролитической диссоциации вскрывает наиболее уязвимые места теории Аррениуса.