
7.3. Контрольно-разрешительная система на отраслевом рынке








7.3.1. Понятие, задачи и структура контрольно-разрешительной системы
в сфере обращения лекарственных средств
Контрольно-разрешительная система в сфере фармацевтической деятельности — это комплекс мероприятий по обеспечению качества лекарственных средств, изделий медицинского назначения, медицинской техники, биологически активных добавок, лечебно-профилактических средств, косметических и стоматологических товаров. В более широком смысле контрольно-разрешительная система является частью (подсистемой) системы государственного регулирования фармацевтической деятельности.
Главной задачей контрольно-разрешительной системы в сфере обращения ЛС (КРС) является защита потребителей от негативных последствий применения лекарств, связанных с их недостаточной изученностью на этапе разрешения и внедрения в фармацевтический оборот, выпуском отечественными фармпроизводителями или ввозом в страну из-за рубежа недоброкачественных лекарственных средств, нарушениями условий их хранения и реализации.
Структура контрольно-разрешительной системы в сфере обращения ЛС включает в себя следующие элементы:
♦ государственную регистрацию лекарственных средств;
♦ техническое регулирование и сертификацию лекарственных средств;
♦ государственный контроль качества, эффективности и безопасности лекарственных средств.
Действие КРС происходит на нескольких уровнях: федеральном, межрегиональном (в границах федеральных округов), региональном (в границах субъекта РФ) и производственном (в организациях-фарм-производителях).
7.3.2. Государственная регистрация лекарственных средств
Согласно пункту 1 статьи 19 Федерального закона от 22 июня 1998 г. № 86-ФЗ, лекарственные средства могут производиться, продаваться и применяться на территории РФ, только если они зарегистрированы федеральным органом исполнительной власти, в компетенцию которого входит осуществление государственного контроля и надзора, в сфере обращения ЛС. В настоящее время таким органом является Росздравнадзор России.
Подлежат государственной регистрации также ЛС, предназначенные для лечения животных. Государственная регистрация наркотических средств и психотропных веществ, применяемых в медицине в качестве ЛС и подлежащих государственному контролю, сопровождается внесением указанных средств и веществ в соответствующие списки в порядке, определенном Федеральным законом «О наркотических средствах и психотропных веществах». Государственной регистрации подлежат:
♦ новые ЛС;
♦ новые комбинации зарегистрированных ранее ЛС;
♦ ЛС, зарегистрированные ранее, но произведенные в других лекарственных формах, с новой дозировкой или другим составом вспомогательных веществ;
♦ воспроизведенные ЛС (дженерики).
Не подлежат государственной регистрации лекарственные средства, изготовляемые в аптеках по рецептам врачей. Также допускается применение незарегистрированных лекарственных средств при клинических исследованиях и испытаниях лекарственных средств, предназначенных для лечения животных.
Не допускается государственная регистрация различных ЛС под одинаковым названием, а также многократная государственная регистрация одного и того же ЛС под одним или различными названиями.
Государственная регистрация лекарственного средства проводится Росздравнадзором в срок, не превышающий 6 месяцев со дня подачи заявления. Заявление о государственной регистрации лекарственного средства подается заявителем, в качестве которого может выступать организация-разработчик или другое юридическое лицо по ее поручению.
Вместе с заявлением в Росздравнадзор для государственной регистрации лекарственного средства представляется следующий пакет документов:
♦ квитанция об оплате государственной регистрации ЛС;
♦ юридический адрес производителя ЛС;
♦ названия лекарственного средства, включая международное непатентованное название, научное название на латинском языке, основные синонимы;
♦ оригинальное название ЛС, если оно зарегистрировано как торговый знак;
♦ перечень и количество компонентов, входящих в состав ЛС;
♦ инструкция по применению лекарственного средства;
♦ сертификат качества ЛС;
♦ данные о производстве ЛС, первоначальный текст фармакопейной статьи;
♦ методы контроля качества ЛС;
♦ результаты доклинических, фармакологических, токсикологических и клинических исследований ЛС;
♦ результаты ветеринарных исследований, если регистрируется ЛС, предназначенное для лечения животных;
♦ образцы лекарственного средства для проведения экспертизы его качества;
♦ предложения по цене лекарственного средства;
♦ документы, подтверждающие регистрацию лекарственного средства, если оно зарегистрировано вне пределов РФ.
Если регистрируется воспроизведенное ЛС, эквивалентное уже зарегистрированному в РФ оригинальному ЛС, возможно произведенному по другой технологии или с другим составом вспомогательных веществ, Росздравнадзор может применить ускоренную процедуру государственной регистрации.
Зарегистрированное лекарственное средство заносится в Государственный реестр лекарственных средств, а для подтверждения факта государственной регистрации заявителю выдается регистрационное удостоверение.
7.3.3. Техническое регулирование производства и сертификация
лекарственных средств
Техническое регулирование производства и сертификация лекарственных средств осуществляются с целью установления их соответствия стандартам качества.
Стандартизация определяется в литературе [106, с. 135] как деятельность по установлению норм, правил и характеристик, отражающих требования государства к безопасности и качеству товаров и создающих необходимое нормативное поле, которым можно руководствоваться производителям и потребителям продукции и услуг.
В области стандартизации лекарственных средств в 1998-2003 гг. был разработан и введен в действие ряд нормативных документов, соответствующих мировым стандартам. Это Правила организации качественных клинических исследований (GCP), Правила организации производства и контроля качества лекарственных средств (GMP), Правила оптовой торговли лекарственными средствами (GDP), а также Правила отпуска (реализации) лекарственных средств в аптечных организациях (GPP).
Приказом Минздрава РФ от 26 марта 2001 г. № 88 введен в действие Государственный информационный стандарт лекарственных средств (ГИСЛС), который устанавливает требования и структуру официальной информации о ЛС. Он состоит из следующих элементов: фармакопейной статьи, формулярной статьи, клинико-фармакологической статьи и паспорта ЛС.
Формулярная статья — это нормативный документ, содержащий cтандарти-зированные по форме и содержанию сведения о применении ЛС при определенном заболевании.
Клинико-фармакологическая статья подразделяется на:
♦ типовую клинико - фармакологическую статью лекарственного средства (ТКФС) — официальный документ, содержащий сведения об основных свойствах ЛС или часто используемых его комбинаций, определяющих эффективность и безопасность, разрабатываемый экспертным органом и утверждаемый Росздравнадзором;
♦ клинико-фармакологическую статью лекарственного препарата (КФС) — официальный документ, отражающий совокупность клинико-фармакологических данных, характеризующих эффективность и безопасность конкретного лекарственного препарата с торговым наименованием, разрабатываемый на основе ТКФС. Проект КФС представляется организацией-фармпроизводителем на экспертизу и утверждается Росздравнадзором.
Паспорт лекарственного средства представляет собой официальный документ, содержащий обобщенную информацию о лекарственном препарате, имеющий юридическое значение в сфере обращения ЛС, в том числе идентифицирующую отличительные свойства упаковки.
На основе ГИСЛС разрабатываются такие нормативные документы, как: Государственный реестр лекарственных средств; перечень жизненно необходимых и важнейших ЛС; список льготного отпуска ЛС; список ЛС, отпускаемых без рецепта врача; обязательный ассортимент ЛС для аптечных организаций, обслуживающих амбулаторных больных; Федеральное руководство для врачей по использованию лекарственных средств.
Приказом Минздрава России от 01 ноября 2001 г. № 388 утвержден ОСТ 91500.05.001—2000 «Стандарт качества лекарственных средств. Основные положения», установивший порядок разработки, оформления, экспертизы, согласования, утверждения, присвоения обозначения и регистрации государственных стандартов качества Л С. Действие этого стандарта распространяется на готовые лекарственные средства отечественного производства.
Государственные стандарты качества ЛС разрабатываются и утверждаются в следующих видах: общая фармакопейная статья; фармакопейная статья; фармакопейная статья на Л С конкретной организации-производителя.
Общая фармакопейная статья (ОФС) включает в себя перечень нормируемых показателей или методов испытания для конкретной лекарственной формы, описание физических, физико-химических, химических, биохимических, биологических и микробиологических методов анализа ЛС, требования к используемым реактивам, титрованным растворам и индикаторам.
Фармакопейная статья (ФС) разрабатывается на лекарственное средство иод международным непатентованным названием, если оно имеется (для монокомпонентных ЛС), и содержит обязательный перечень показателей и методов контроля качества (с учетом его лекарственной формы), соответствующих положениям ведущих зарубежных фармакопеи. Разработка фармакопейной статьи на оригинальное (патентованное) ЛС в течение срока действия патентной защиты и включение ее в Государственную фармакопею возможна лишь по согласованию с разработчиком ЛС либо осуществляется после окончания срока действия патента.
Общие и специальные фармакопейные статьи составляют Государственную фармакопею, которая издается Росздравом и подлежит переизданию каждые 5 лет.
Фармакопейная статья на ЛС конкретной организации-производителя (ФСП) содержит перечень показателей и методов контроля качества ЛС производства конкретной организации и разрабатывается с учетом требований Государственной фармакопеи и стандартов. Требования к качеству лекарственных средств, содержащиеся в ФСП, должны быть не ниже требований, изложенных в Государственной фармакопее, с учетом требований стандарта качества лекарственных средств № 388. Срок действия ФСП устанавливается с учетом уровня технологического процесса конкретного производства лекарственного средства, но не более 5 лет. При разработке нового ЛС в случае отсутствия государственного стандарта качества ЛС на субстанцию одновременно с разработкой ФСП на лекарственный препарат разрабатывается ФСП на субстанцию, используемую для его производства.
Фармакопейные статьи, то есть государственные стандарты ЛС, содержащие перечень показателей и методов контроля качества, призваны обеспечивать разработку качественного, эффективного и безопасного ЛС, для чего должны своевременно пересматриваться с учетом новых достижений медицинской, фармацевтической и других наук и положений ведущих зарубежных фармакопеи, рекомендаций ведущих международных организаций в области фармацевтической науки.
Основным инструментом обеспечения качества лекарственной продукции в товаропроизводящем звене в настоящее время является сертификация.
Сертификация лекарственных средств — это форма подтверждения соответствия лекарственных средств требованиям нормативных документов (НД) специально аккредитованными органами. Все серии зарегистрированных в России лекарственных средств, выпускаемых организациями-производителями или ввозимых из-за рубежа, подлежат сертификации. Реализация ЛС без сертификата запрещена.
Правовое регулирование сертификации и технического регулирования производства лекарственных средств осуществляются в настоящее время Законом РФ «О защите прав потребителей» от 07 февраля 1992 г. № 2300-1, Федеральным законом «О техническом регулировании» от 27 декабря 2002 г. № 184, постановлением Правительства РФ «Об утверждении перечня товаров, подлежащих обязательной сертификации, и перечня работ и услуг, подлежащих обязательной сертификации» от 13 августа 1997 г. № 1013, а также Правилами проведения сертификации в Системе сертификации лекарственных средств Системы сертификации ГОСТ Р, утвержденными постановлением Госстандарта РФ от 24 мая 2002 г. № 36.
Система сертификации Л С представлена следующими участниками:
♦ Центральный орган Системы (ДО) — Управление государственного контроля лекарственных средств и медицинской техники Рос-здрава России;
♦ Органы сертификации лекарственных средств (ОС) — аккредитованные ГОСТ Р органы, возглавляющие работу но сертификации ЛС на подведомственной им территории. По состоянию на декабрь 2002 г. на территории России было аккредитовано 8 ОС: Центр сертификации Росздрава России и 7 центров сертификации в федеральных округах (в Москве, Санкт-Петербурге, Нижнем Новгороде, Ростове-на-Дону, Екатеринбурге, Новосибирске и Хабаровске);
♦ Испытательные лаборатории (ИЛ) — лаборатории, аккредитованные ГОСТ Р на проведение сертификационных испытаний ЛС на соответствие нормативным документам, утвержденным Минздравсоцразвитием РФ (ОФС, ФС, ФСП, НД на ЛС зарубежного производства).
При сертификации ЛС применяются схемы сертификации, принятые в Порядке проведения сертификации продукции в РФ, утвержденном постановлением Госстандарта РФ от 21 сентября 1994 г. № 15:
♦ схема № 5 — сертификация производства или системы качества, контроль системы качества (производства), испытание образцов, взятых у продавца или изготовителя. Данная схема может применяться при сертификации ЛС, произведенных организациями, имеющими сертификат соответствия систем качества (производства);
♦ схема № 7 — испытание партии (серии) ЛС.
Выбор объема проводимых испытаний в каждом конкретном случае определяется органом по сертификации. При сертификации ЛС изучается информация о продукции, нормативных документах, регламентирующих показатели и методы испытаний; проводится идентификация продукции, в том числе проверяется происхождение и соответствие сопроводительной и нормативной документации, принадлежность к данной партии. Для подтверждения соответствия ЛС требованиям, установленным нормативными документами, проводятся испытания их характеристик (показателей).
Участники Системы, виновные в нарушении правил обязательной сертификации ЛС, несут в соответствии с действующим законодательством уголовную, административную либо гражданско-правовую ответственность.
7.3.4. Государственный контроль качества, эффективности и
безопасности лекарственных средств
Вопросы качества, эффективности и безопасности лекарственных средств в настоящее время находятся в центре внимания законодателя. Статья 4 Федерального закона «О лекарственных средствах» определяет основные понятия, связанные с качеством и безопасностью лекарственных средств.
Качество лекарственных средств определяется как соответствие лекарственных средств государственному стандарту их качества. Документом, подтверждающим соответствие качества лекарственного средства государственному стандарту, является сертификат качества лекарственного средства.
Безопасность означает характеристику лекарственных средств, основанную на сравнительном анализе их эффективности и оценке риска причинения вреда здоровью. Эффективность лекарственных средств, в свою очередь, означает характеристику степени положительного влияния на течение болезни.
Фальсифицированное лекарственное средство определено как ЛС, сопровождаемое ложной информацией о составе или производителе лекарственного средства.
Понятие недоброкачественного лекарственного средства подразумевает негодность или истечение срока годности ЛС.
Согласно статье 8 Федерального закона «О лекарственных средствах» государственному контролю подлежат все лекарственные средства, произведенные на территории РФ и ввозимые из-за рубежа. Порядок осуществления государственного контроля качества лекарственных средств на территории РФ регламентируется в настоящее время приказом Министерства здравоохранения РФ от 04 апреля 2003 г. № 137.
Данным нормативным актом предусмотрены следующие виды государственного контроля качества лекарственных средств: предварительный, выборочный и повторный выборочный, контроль качества фармацевтических субстанций, а также периодические проверки производителей с целью инспектирования качества выпускаемых ими ЛС.
Предварительному контролю качества подлежат следующие ЛС: впервые производимые организацией на территории РФ; впервые ввозимые на территорию РФ из-за рубежа; выпускаемые по измененной технологии; выпускаемые после перерыва более 3 лет; в связи с ухудшением их качества.
Процедура предварительного контроля качества включает в себя следующие этапы:
♦ направление организацией-производителем в Росздравнадзор России заявки с пакетом необходимых документов;
♦ анализ документов и выдача Росздравнадзором разрешения на проведение предварительного контроля качества;
♦ отбор образцов для целей предварительного контроля качества;
♦ направление образцов на экспертизу качества;
♦ проведение экспертизы качества представленных образцов;
♦ принятие Росздравнадзором решения по результатам проведенной экспертизы качества.
Для получения решения о направлении на предварительный контроль качества ЛС организация-производитель направляет в Росздравнадзор вместе с заявкой следующий пакет документов: заверенную копию регистрационного удостоверения; заверенные копии титульных листов государственного стандарта качества и согласованного в установленном порядке технологического регламента производства; копию аттестата контрольных лабораторий отдела контроля качества (ОКК) организации-производителя на техническую компетентность в области контроля качества производимых ЛС.
Решение о направлении на проведение предварительного контроля выдается Росздравнадзором в течение 20 рабочих дней с даты поступления заявки и документов.
Организации-производители, впервые начинающие серийный выпуск ЛС, должны направить на предварительный контроль качества образцы первых 3 промышленных серий этого ЛС по мере их наработки. Если предварительный контроль качества проводится в связи с ухудшением его качества, контролю подлежат 5 очередных серий ЛС.
Организации, осуществляющие упаковку или расфасовку ЛС, созданных другими производителями (отечественными или зарубежными), направляют на предварительный контроль качества образцы трех промышленных серий упакованного или расфасованного ЛС.
При изменении наименования лекарственного средства организация-производитель направляет на предварительный контроль качества одну серию переименованного лекарственного средства.
Экспертиза качества лекарственного средства проводится в срок, не превышающий 30 рабочих дней со дня поступления образцов и комплекта документов, если в государственном стандарте качества не предусмотрены методы, требующие более длительных сроков выполнения экспертизы. По окончании экспертизы качества ее результаты с протоколом анализа направляются в Росздравнадзор России и организации-производителю.
Лекарственное средство снимается с предварительного контроля качества и переводится на выборочный, если качество всех представленных образцов соответствует требованиям государственного стандарта качества для него.
На основании решения Росздравнадзора о снятии ЛС с предварительного контроля качества, а также по результатам экспертизы эффективности и безопасности Росздравнадзором оформляется решение о выпуске ЛС в обращение на территории РФ. Данное решение является основанием для выдачи организации-производителю заключения Росздравнадзора о соответствии организации производства ЛС требованиям Федерального закона «О лекарственных средствах».
При наличии замечаний к качеству лекарственного средства по результатам проведения экспертизы качества оно не подлежит снятию с предварительного контроля.
Выборочному контролю качества подлежат лекарственные средства отечественного и зарубежного производства, находящиеся в обращении в РФ.
Номенклатура и периодичность отбора образцов на выборочный контроль качества регламентируются планом выборочного контроля, который доводится до сведения организаций-производителей в виде планов-заданий. В течение календарного года план-задание может корректироваться с учетом изменения номенклатуры ЛС, находящихся в обращении в РФ, или выявленного несоответствия их качества требованиям государственных стандартов качества.
Процедура выборочного контроля качества ЛС включает следующие этапы:
♦ принятие Росздравнадзором решения о проведении выборочного контроля качества ЛС в соответствии с утвержденным планом;
♦ отбор образцов для целей выборочного контроля качества;
♦ направление образцов на экспертизу качества;
♦ проведение экспертизы качества представленных образцов;
♦ принятие Росздравнадзором решения по результатам проведенной экспертизы качества.
Экспертиза качества проводится в срок, не превышающий 40 рабочих дней со дня получения образцов ЛС и комплекта документов, если в государственном стандарте качества не предусмотрены методы контроля, требующие более длительных сроков их выполнения. Результаты экспертизы в рамках выборочного контроля качества ЛС направляются в Росздравнадзор и организации-производителю.
При выявлении несоответствия качества ЛС требованиям государственного стандарта Росздравнадзор направляет информацию об изъятии партии некачественного ЛС в территориальные органы контроля качества, которые принимают меры к выявлению и изъятию его из обращения.
Выборочный контроль качества сертифицированных лекарственных средств, находящихся в обращении на территории РФ, при поступлении их по месту назначения осуществляется территориальными органами контроля качества в рамках инспекционного контроля. В ходе инспекционной проверки проводится выборочный контроль качества по показателям «описание», «упаковка», «маркировка», проверяется происхождение, соответствие сопроводительной документации и государственному стандарту качества, принадлежность к данной партии ЛС.
При возникновении сомнений в достоверности данных, полученных в результате проверки сопроводительной документации и контроля качества ЛС, проводятся дополнительные испытания. Их объем для выборочной проверки в каждом конкретном случае определяется территориальным органом контроля качества. Территориальный орган контроля качества лекарственных средств представляет в Росздравнадзор информацию о случаях выявления несоответствия качества ЛС требованиям государственных стандартов, незаконных копий или подделок зарегистрированных в РФ лекарственных средств, а также направляет ежемесячный отчет о результатах проверок качества сертифицированных ЛС.
Повторному выборочному контролю качества подлежат ЛС в случае возникновения споров об их качестве между субъектами обращения. Он проводится по решению Росздравнадзора России.
Процедура повторного выборочного контроля включает следующие этапы:
♦ направление организацией-производителем в Росздравнадзор заявки на повторный выборочный контроль качества ЛС и необходимых документов с обоснованием его проведения;
♦ рассмотрение Росздравнадзором представленных документов и принятие решения о проведении повторного выборочного контроля качества ЛС;
♦ отбор образцов ЛС для повторного выборочного контроля их качества;
♦ направление образцов на экспертизу качества;
♦ проведение экспертизы качества представленных образцов;
♦ принятие Росздравнадзором решения по представленным результатам экспертизы качества.
На повторный выборочный контроль качества лекарственные средства могут направляться предприятием, выявившим их несоответствие требованиям государственных стандартов качества, а также организацией-производителем, направляющей на повторный выборочный контроль архивные образцы данного ЛС.
Решение о проведении повторного выборочного контроля качества лекарственного средства принимается Росздравнадзором в течение 20 рабочих дней с даты поступления заявки и документов.
Экспертиза качества лекарственного средства для целей повторного выборочного контроля проводится в срок не более 20 рабочих дней со дня получения образцов и комплекта документов, если в государственном стандарте качества не предусмотрены методы контроля, требующие более длительных сроков.
Результаты экспертизы качества ЛС с протоколом анализа направляются в Росздравнадзор России и субъектам обращения, представившим образцы на повторный выборочный контроль качества.
Государственный контроль качества фармацевтических субстанций проводится на этапе их регистрации, а также в рамках предварительного и выборочного контроля их качества как лекарственных средств на основании решения Росздравнадзора. Экспертизе качества подлежат все субстанции на этапе их регистрации, а также субстанции, ввозимые на территорию РФ и предназначенные для изготовления лекарственных средств.
Экспертиза качества субстанций проводится в срок не более 30 рабочих дней со дня получения образцов субстанции и стандартных образцов веществ, необходимых для проведения экспертизы качества, если в государственном стандарте не предусмотрены методы контроля, требующие более длительных сроков. Разъяснения вопросов, связанных с контролем субстанций, содержатся в письме Департамента государственного контроля лекарственных средств и медицинской техники Минздрава России от 04 марта 2004 г. № 295-22/37.
Образцы ЛС для проведения предварительного, выборочного и повторного выборочного контроля качества отбираются специалистами Росздравнадзора или учреждений, входящих в государственную систему контроля качества, эффективности и безопасности.
Отбор образцов отечественных ЛС для предварительного и выборочного контроля качества осуществляется из архивных образцов лекарственных средств ОКК организации-производителя. Отбор образцов на выборочный контроль качества может проводиться также при проверках организации-производителя. Отбор образцов зарубежных ЛС на выборочный контроль качества проводится со складов на территории РФ, указанных зарубежной организацией-производителем.
Порядок направления образцов на государственный контроль качества лекарственных средств. Образцы лекарственных средств направляются на государственный контроль качества в упаковке, предусмотренной государственным стандартом; образцы субстанций — в таре из стекла. Маркировка образцов лекарственных средств должна соответствовать требованиям государственных стандартов качества.
На предварительный и выборочный контроль качества и контроль качества субстанции на этапе регистрации образцы направляются в количестве, достаточном для проведения трех анализов в соответствии с требованиями государственного стандарта качества (с учетом испытания па микробиологическую чистоту). Образцы ЛС для инъекций и глазных капель направляются с учетом испытаний показателя «механические включения», а образцы лекарственного растительного сырья — с учетом результатов радиационного контроля.
Образцы ЛС, направляемых на предварительный или выборочный контроль качества, а также образцы субстанций должны сопровождаться стандартными образцами субстанций и веществ, необходимых для проведения контроля в соответствии с государственными стандартами качества. Образцы субстанции направляются в количестве, достаточном для проведения двух анализов.
На выборочный контроль качества образны отечественных ЛС направляются с архивным образцом субстанции в количестве, достаточном для проведения двух анализов в соответствии с утвержденным государственным стандартом.
На повторный выборочный контроль качества ЛС организация-производитель направляет образцы в ненарушенной упаковке. Количество упаковок, составляющих образец, рассчитывается по формуле: 0,4 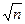 , где п — количество упаковок в одной серии, но не более 30. Количество образцов ЛС, направляемых на повторный выборочный контроль качества по показателям «механические включения» и «радиационный контроль», определяется соответствующими государственными стандартами качества.
, где п — количество упаковок в одной серии, но не более 30. Количество образцов ЛС, направляемых на повторный выборочный контроль качества по показателям «механические включения» и «радиационный контроль», определяется соответствующими государственными стандартами качества.
Образцы ЛС направляются на государственный контроль качества с сопроводительным письмом, в котором указывается вид контроля качества, с сертификатом качества организации-производителя и актом отбора образцов. Образцы для инъекций и глазных капель, направляемые на предварительный контроль качества, кроме вышеуказанных документов должны сопровождаться результатами проверки качества указанных ЛС по показателю «механические включения». Результаты такой проверки представляют территориальные органы контроля качества.
Образцы субстанций, из которых произведены ЛС, представляемые на предварительный и выборочный контроль качества (для отечественных организаций-производителей), должны сопровождаться сертификатом качества, выданным по результатам контроля качества субстанции при ее поступлении в производство по всем показателям, оригиналом или заверенной копией сертификата качества ЛС; а для зарубежных субстанций дополнительно должны быть указаны даты изготовления и окончания срока годности.
Образцы, оставшиеся после проведения государственного контроля качества лекарственных средств, хранятся не менее 6 месяцев, после чего образцы, не удовлетворяющие требованиям государственного стандарта качества, подлежат уничтожению. Образцы ЛС, удовлетворяющие требованиям государственных стандартов качества, возвращаются организациям-производителям по их письменной просьбе либо используются в научно-исследовательских целях или безвозмездно передаются в учреждения здравоохранения по их письменным заявкам.
7.4. Правовые основы защиты информации, государственной,
коммерческой и служебной тайны
7.4.1. Правовые основы защиты информации
В отечественном законодательстве информация определяется как сведения о лицах, предметах, фактах, событиях, явлениях и процессах независимо от формы их представления. Правовой основой защиты информации является Федеральный закон «Об информации, информатизации и защите информации» от 20 февраля 1995 г. № 24-ФЗ. Данным законом в оборот вводятся следующие термины: «документированная информация», «конфиденциальная информация», «персональные данные граждан».
Документированной информацией (документом) является зафиксированная на материальном носителе информация с реквизитами, позволяющими ее идентифицировать. Документирование информации является обязательным условием включения информации в информационные ресурсы. На гражданах, юридических лицах, органах государственной власти и местного самоуправления лежит обязанность предоставлять документированную информацию органам и организациям, ответственным за формирование и использование государственных информационных ресурсов.
Государственные информационные ресурсы РФ являются открытыми и общедоступными. Исключение составляет документированная информация, отнесенная законом к категории ограниченного доступа.
Документированная информация с ограниченным доступом по условиям ее правового режима подразделяется на информацию, отнесенную к государственной тайне, и конфиденциальную.
Конфиденциальная информация — это документированная информация, доступ к которой ограничивается в соответствии с законодательством РФ. Порядок обязательного предоставления (получения) информации, отнесенной к государственной тайне, и конфиденциальной информации устанавливается и осуществляется в соответствии с законодательством об этих категориях информации1.
При этом статья 10 Федерального закона «Об информации, информатизации и защите информации» запрещает относить к информации с ограниченным доступом:
♦ законодательные и другие нормативные акты, устанавливающие правовой статус органов государственной власти, местного самоуправления, организаций, общественных объединений, а также права, свободы и обязанности граждан, порядок их реализации;
♦ документы, содержащие информацию о чрезвычайных ситуациях, экологическую, метеорологическую, демографическую, санитарно-эпидемиологическую и другую, необходимую для обеспечения безопасного функционирования населенных пунктов, производственных объектов, безопасности граждан и населения в целом;
♦ документы, содержащие информацию о деятельности органов государственной власти и местного самоуправления, использовании бюджетных средств и других государственных и местных ресурсов, состоянии экономики и потребностях населения, за исключением сведений, отнесенных к государственной тайне;
♦ документы, накапливаемые в открытых фондах библиотек и архивов, информационных системах органов государственной власти и местного самоуправления,
♦ общественных объединений, организаций, представляющих общественный интерес, или необходимые для реализации прав, свобод и обязанностей граждан.
Информация о гражданах (персональные данные) — это сведения о фактах, событиях и обстоятельствах жизни гражданина, позволяющие идентифицировать его личность.
Персональные данные относятся к конфиденциальной информации. Не допускается сбор, хранение, использование и распространение информации о частной жизни, а равно нарушающей личную, семейную тайну, тайну переписки, телефонных переговоров, почтовых, телеграфных и иных сообщений физического лица без его согласия, кроме как на основании судебного решения. Персональные данные не могут быть использованы в целях причинения имущественного и морального вреда гражданам, затруднения реализации прав и свобод граждан РФ. Ограничение прав граждан Российской Федерации на основе использования информации об их социальном происхождении, расовой, национальной, языковой, религиозной и партийной принадлежности запрещено и карается законом.
Защите подлежит любая документированная информация, неправомерное обращение с которой может нанести ущерб ее собственнику, владельцу, пользователю и иному лицу.
 |
1 Например, приказом Министерства по налогам и сборам РФ от 03 октября 2000 г. № БГ-3-24/346 утвержден Порядок предоставления налоговыми органами конфиденциальной информации.
Защита прав субъектов в сфере формирования информационных ресурсов, пользования информационными ресурсами, разработки, производства и применения информационных систем, технологий и средств их обеспечения осуществляется в целях предупреждения правонарушений, пресечения неправомерных действий, восстановления нарушенных прав и возмещения причиненного ущерба. Целями защиты информации являются:
♦ предотвращение утечки, хищения, утраты, искажения, подделки информации;
♦ предотвращение угроз безопасности личности, общества и государства;
♦ предотвращение несанкционированных действий по уничтожению, модификации, искажению, копированию, блокированию информации, других форм незаконного вмешательства в информационные ресурсы и системы, обеспечение правового режима документированной информации как объекта права собственности;
♦ защита конституционных прав граждан на сохранение личной тайны и конфиденциальности персональных данных, имеющихся в информационных системах;
♦ сохранение государственной тайны и конфиденциальности документированной информации;
♦ обеспечение прав субъектов в информационных процессах и при разработке, производстве и применении информационных систем, технологий и средств их обеспечения.
Права и обязанности субъектов в области защиты информации установлены статьей 22 Федерального закона «Об информации, информатизации и защите информации». Федеральный закон закрепляет защиту прав на доступ к информации. Отказ в доступе к открытой информации или предоставление пользователям заведомо недостоверной информации могут быть обжалованы в судебном порядке. Во всех случаях лица, которым отказано в доступе к информации, и лица, получившие недостоверную информацию, имеют право на возмещение понесенного ими ущерба.