
Одностадийная технология.








В данном варианте технологии окисление этилена и полная регенерация каталитической системы совмещены в одном реакторе (1) (Рис. 4.7).
Температура процесса в реакторе 130 оС, давление около 30 атм. Реактор (1) представляет собой пустотелую барботажную колонну. В низ реактора подают свежий и рециркулируемый этилен и кислород. Тепло экзотермической реакции отводится за счет испарения реакционной массы и конденсации ее паров в холодильнике (2). Во избежание создания взрывоопасных смесей органики с кислородом используют избыток этилена по отношению к стехиометрии реакции, чтобы достичь в реакторе полной конверсии кислорода. Избыточный этилен рециркулируют, что заставляет использовать реагенты высокой чистоты (кислород с концентрацией 99% и этилен с концентрацией 99,7%), чтобы минимизировать сдувку рециркулирующего газа.
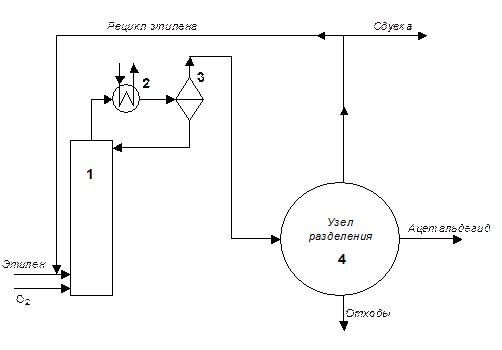
Рис. 4.7. Схема реакторного узла одностадийной технологии окисления этилена до ацетальдегида.
Образующийся ацетальдегид с верха реактора постоянно выдувается потоком избыточного этилена. Конденсируемые в холодильнике (2) жидкие компоненты отделяются от газов в сепараторе (3) и возвращаются в реактор. Газообразный поток, содержащий этилен, ацетальдегид и примеси, из сепаратора направляют в узел разделения (4). Преимуществом данного метода является отсутствие циркуляции катализатора (комплекс палладия, растворенный в жидкой реакционной массе не покидает реактор), что упрощает задачу минимизации потерь палладия.
Сравнение одно- и двух-стадийной технологий.
Краткое сравнение преимуществ и недостатков рассмотренных методов приведено в Таблице 4.3. Высокая кислотность реакционной массы заставляет использовать дорогие кислотостойкие материалы для изготовления аппаратуры. В случае одностадийного способа это касается только реактора, а в случае двухстадийного - всех аппаратов, по которым циркулирует раствор палладия. С точки зрения взрывобезопасности преимущество имеет двухстадийная технология, т.к. по ней кислород не контактирует ни с этиленом, ни с ацетальдегидом. Требования к чистоте сырья выше в одностадийном методе, что определяет бόльшие материальные затраты на реагенты. Проблема минимизации потерь палладия решается проще в одностадийном способе.
В каждом конкретном случае выбор способа производства определяется по экономическим критериям. В настоящее время соотношение установок, работающих по одно- и двух-стадийной технологии составляет примерно 15/85.
Таблица 4.3.
Сравнение одно- и двух-стадийной технологий окисления этилена до ацетальдегида.
| Процесс | Дорогие материалы аппаратуры | Взрыво-безопасность | Температура, давление | Стоимость сырья | Простота минимизации потерь палладия |
| Одностадийный | + (только реактор) | - (О2 и этилен контактируют в одном аппарате) |
Примерно одинаковые | - (О2 и этилен высокой чистоты) | + (палладий всегда в одном аппарате) |
| Двухстадийный | - (все аппараты) | +(О2 и этилен не контактируют) | + (воздух) | - (палладий циркулирует через три аппарата) |
4.5. ОКИСЛЕНИЕ ЦИКЛОГЕКСАНА (ПРОИЗВОДСТВО ЦИКЛОГЕКСАНОЛА И ЦИКЛОГЕКСАНОНА)
Механизм рассмотренного выше Вакер-процесса нетипичен для реакций окисления органических соединений кислородом. Как правило реакции окисления протекают по радикально-цепному механизму. Одним из основных продуктов такого окисления являются гидропероксиды, которые, будучи высоко реакционно-способными, могут претерпевать дальнейшие превращения давая другие продукты.
Одним из важных промышленных процессов, протекающих по радикально-цепному механизму является окисление циклогексана до циклогексанола (анола) и циклогексанона (анона). Схема реакции выглядит так:
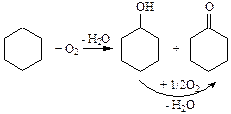 (4.18)
(4.18)
В качестве катализаторов этой реакции применяют соли металлов переменной валентности. Роль металлов-катализаторов заключается в участии в окислительно-восстановительных реакциях генерации радикалов на стадии зарождения цепи и реакциях каталитического разложении гидроперексидов до радикалов.
В промышленности чаще всего используют растворимые в органике соли Со и Mn.
Механизм реакции.
Радикально-цепной механизм сложен и приводит к образованию большого количества побочных продуктов окислении, так как кислород обладает высокой активностью по отношению реагентам, промежуточным и целевым продуктам. Упрощенную схема только целевой каталитической реакции можно представить следующим образом:
зарождение цепи:
R-H + Co3+ à R· + H+ + Co2+ (4.19)
развитие цепи и образование продуктов:
(4.20)
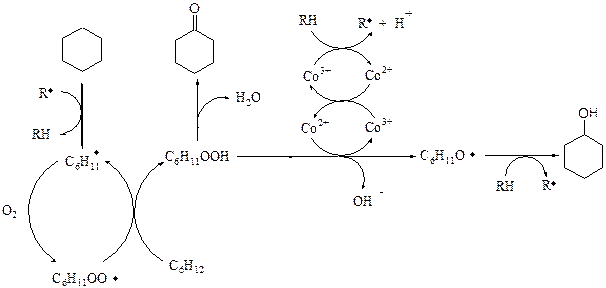
Зарождение цепи протекает по окислительно-восстановительной реакции (4.19) с участием Со3+ и молекулы углеводорода (например циклогенксана). При этом генерируется радикал (R·), а катион кобальта восстанавливается на единицу заряда.
Далее идет развитие цепного процесса (4.20). Радикал (R·) отрывает атом водорода от молекулы циклогексана, образуя соответствующий радикал (С6Н11·), который взаимодействует с молекулой кислорода, давая пероксо-радикал. Последний, в свою очередь, отрывает атом водорода от новой молекулы циклогексана, превращаясь в гидропероксид циклогексана и генерируя новый циклогексил-радикал (так происходит развитие цепи).
Образование целевых продуктов происходит в результате химических превращений гидропероксида циклогексана. Циклогексанон образуется по реакции некаталитического распада гидропероксида. Параллельно гидропероксид взаимодействует с катионами кобальта. Целевым здесь является окисление Со2+ до Со3+ с образованием радикала С6Н11О·, который далее отрывая атом водорода от RH превращается в циклогексанол.
Таким образом кобальт включен в циклический процесс окисления-восстановления, поэтому действует как катализатор и добавляется в реакционную массу в очень небольших концентрациях.
Описание работы реакционного узла.
Схема реакционного узла представлена на Рисунке 4.8. Процесс проводят в одной или в каскаде полых барботажных колонн (1) при температуре 125-165 оС и давлении в 8-15 атм.
Для поддержания высокой селективности образования целевых продуктов (80-85%) конверсию циклогексана ограничивают 10-12 %-ами. В результате побочных реакций окисления образуется целый ряд нежелательных продуктов более глубокого окисления (кислоты, лактоны, кетоспирты, СО2 и пр.). Соотношение образующихся анола:анона примерно составляет 2:3.
Тепло экзотермической реакции снимается кипением циклогексана, который конденсируется в холодильнике (2) и возвращается в процесс, смешиваясь со свежим потоком циклогексана. Из последнего реактора смесь попадает в экстрактор (3), где обрабатывается водой. Соли кобальта переходят в водный раствор, который отделяется от органического слоя в сепараторе (4). Органический слой после сепаратора отправляют в узел разделения (5), где отгоняют избыточный циклогексан, возвращая его в процесс, и выделяют чистые анон и анол.
Катализатор в данном процессе применяют в концентрациях около 20 ppm. Учитывая, что цена на кобальт и марганец не высока и концентрации их в реакционной массе низкие, то обычно не организовывают рецикл катализатора. Удаляют соли металлов обработкой водной щелочью и фильтрацией выпадающих в осадок гидроксидов. Альтернативным вариантом может служить поглощение катионов металлов селективными катионо-обменными смолами, с последующей их регенерацией.
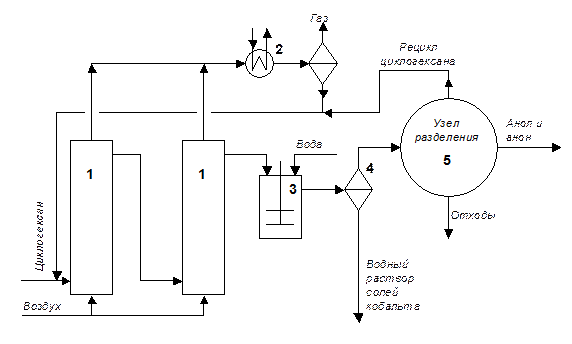
Рис. 4.8. Схема реакционного узла окисления циклогексана.
4.6. ЭПОКСИДИРОВАНИЕ ОЛЕФИНОВ (ХАЛКОН-ПРОЦЕСС)
Компанией “Халкон” был разработан процесс совместного получения стирола (или изобутилена) и оксида пропилена. Халкон-процесс представляет собой комбинацию трех технологических процессов в одном производстве. При "стирольном" варианте процесса сырьем для получения гидропероксида является этилбензол, а при "изобутиленовом" варианте - изобутан. В случае этилбензола процесс включает следующие стадии:
1. Окисление этилбензола до гидроперексида (ГП):
C6H5C2H5 + O2 à C6H5CH(CH3)OOH (4.21)
2. Эпоксидирование пропилена, катализируемое комплексами переходных металлов:
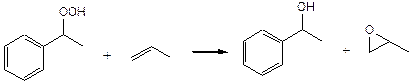 (4.22)
(4.22)
3. Дегидратация образующегося на предыдущей стадии метилфенилкарбинола (МФК) до стирола (гетерогенный кислотный катализ):
C6H5CH(CH3)OH + H2O à C6H5CH=CH2 (4.23)
Вторая стадия Халкон-процесса катализируется комплексами переходных металлов. В качестве катализаторов используют комплексы металлов, являющихся жесткими центрами (MoVI, WVI, TiIV), так как они хорошо активируют кислород гидропероксида, являющегося также жестким центром. В промышленности используют, в большинстве случаев, комплексы молибдена, как самые активные и селективные в данном процессе.
Механизм катализа.
Каталитический цикл реакции эпоксидирования был рассмотрен ранее (п. 3.3.5.2.1. уравнение (3.49)).
Сырье и продукты.
В промышленности по реакции эпоксидирования получают главным образом оксид пропилена, реже - оксид стирола и некоторые другие эпоксиды. Различие процессов состоит в используемом сырье для получения гидропероксида. В основном применяют два углеводорода: этилбензол (с образование, в качестве второго продукта производства, стирола) и изобутан (с образованием изобутилена).
Побочные реакции.
Параллельно целевой реакции эпоксидирования протекает побочная реакция разложения гидроперекиси, в основном с образованием ацетофенона:
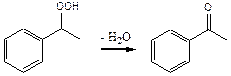 (4.24)
(4.24)
Селективность образования оксида пропилена по исходному пропилену составляет 95-97%, а по гидроперексиду – 80-85%.
Описание работы реакторного узла.
С целью снижения доли реакции разложения гидроперекиси и увеличения выхода целевых продуктов, процесс проводят в 2-5 кратном избытке пропилена при мягких условиях: температура 90-110 оС, давление в 20-30 ат. Концентрация катализатора 0,001-0,005 моль/(моль ГП).
Схема реакторного узла представлена на Рисунке 4.9. Процесс проводят в каскаде емкостных реакторов с мешалкой и охлаждением. В первый реактор каскада подают поток пропилена (свежего и рециркулируемого). Туда же подают гидропероксид, представляющий собой 20-30%-ный раствор в исходном этилбензоле, с растворенным в нем катализатором. После каскада реакторов смесь направляется в узел разделения, представляющий собой последовательность ректификационных колонн, где отгоняют избыточный пропилен (возвращаемый в процесс) и выделяют продукты реакции. Катализатор, как нелетучее вещество, остается в кубовых остатках. Часть кубового остатка возвращают в процесс с целью частичной рециркуляции катализатора. Поскольку цена на молибден относительно не высока, а концентрации его в процессе низкие, то организовывать полную регенерацию и рецикл катализатора экономически не выгодно. Из кубовых остатков его можно осаждать и отфильтровывать, либо поглощать селективными комплексообразующими ионитами с последующим выделением из них.
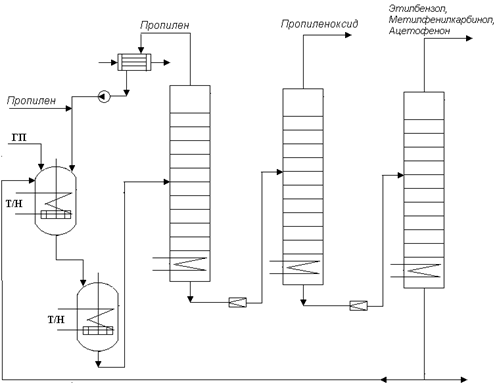
Рис.4.9. Схема реакторного узла процесса эпоксидирования пропилена.
4.7. ОЛИГОМЕРИЗАЦИЯ ЭТИЛЕНА ( SHOP - PROCESS )
Сырье и продукты.
Данный процесс (Shell Higher Olefin Process - SHOP) предназначен для производства высших линейных a-олефинов каталитической олигомеризацией этилена. Получаемые по данной технологии a-олефины являются сырьем в производстве поверхностно-активных веществ. Кроме того, производимые в этом же процессе 1-гексен и 1-октен сополимеризуют с этиленом для получения высокопрочного полиэтилена (для упаковочных материалов). 1-Децен используется для производства высокотемпературных моторных масел.
Химизм и блок-схема процесса.
В основе технологии лежит реакция олигомеризации этилена, катализируемая гомогенными комплексами переходных металлов. Олигомеризацию хорошо катализируют комплексы кобальта, титана и никеля. Компания «Shell» разработала никелевый катализатор с карбоксифосфиновым лигандом:
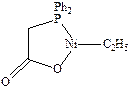
который обеспечивает оптимальное соотношение скоростей стадий внедрения и b-элиминирования (см. механизм катализа п. 3.3.3.) в каталитическом цикле, позволяющее получать смесь олигомеров с узким молекулярно-массовым распределением с максимумом в области степени олигомеризации 6-9. a-Олефины с соответствующим числом углеродов (12-18) являются наиболее ценным сырьем для производства ПАВ.
Блок-схема производства приведена на Рисунке 4.10. Стадию олигомеризации этилена проводят при температуре 80-120 0С и давлении 70-140 атм.
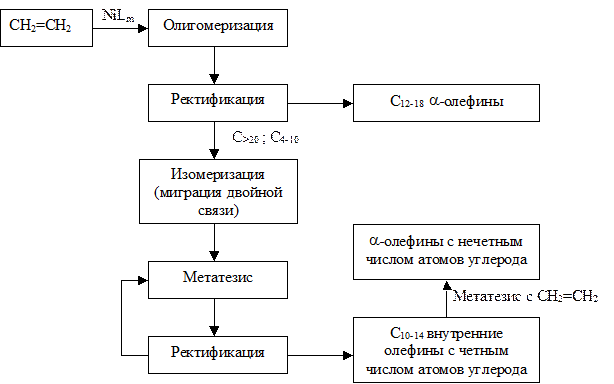
Рис. 4.10. Блок-схема производства линейных a-олефинов олигомеризацией этилена.
Затем олефины разделяют ректификацией на фракции: С4-10, С12-18, С>20. Целевой фракцией при производстве сырья для ПАВ является С12-18. Высшие и низшие фракции смешивают и направляют на стадию каталитической изомеризации с миграцией двойной связи (механизм см. п. 3.3.2.). Получающиеся при этом линейные олефины с внутренней двойной связью подвергают затем реакции диспропорционирования (механизм см. п. 3.3.4.). Химические превращения на этих двух стадиях упрощенно можно представить следующей схемой:
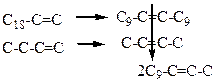 (4.25)
(4.25)
В результате получают смесь олефинов с четным количеством атомов углерода с преобладанием фракции С10-14 и двойной связью в b-положении. Эту фракцию выделяют ректификацией и направляют на стадию диспропорционирования с этиленом (гетерогенно-каталитический процесс с катализатором Re2O7/Sn(CH3)4/Al2O3), в результате которой образуются линейные a-олефины с нечетным числом углеродных атомов, которые также выделяют в качестве товарного продукта. Оставшиеся высшие и низшие олефины рециркулируют.
Каталитический цикл олигомеризации.
Схематично каталитический цикл олигомеризации можно представить следующей схемой:
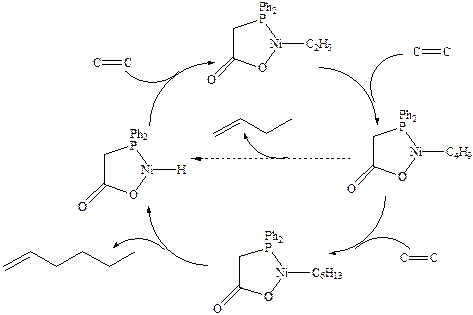 (4.26)
(4.26)
Суммарный выход линейных a-олефинов в данной технологии составляет 94-97 %. Основным преимуществом данного процесса является возможность легко варьировать состав получаемых a-олефинов в зависимости от требований рынка. Благодаря высоким экономическим показателям этот процесс вытесняет используемые ранее способы производства высших a-олефинов: крекингом парафинов и алюминий-органическим синтезом (олигомеризация этилена, катализируемая триэтилалюминием).