
Факторы, влияющие на эффективность катализаторов полимеризации, олигомеризации и димеризации.








Высокой скорости реакции внедрения (r1) способствуют металлы, являющиеся жесткими кислотами (металлы IV-VI групп в высоких степенях окисления: TiIV, TiIII, VIII, CrIII, ZrIV и пр.), так как они способствуют большей поляризации связи металл-олефин оттягивая электронную плотность от олефина и делая его более электрофильным. Это сильно облегчает атаку на олефин нуклеофильным лигандом (гидрид или алкил). Поэтому именно эти металлы используют для каталитической полимеризации a-олефинов и диенов (в частности в производстве полиэтилена и полипропилена).
Одними из наиболее активных катализаторов полимеризации являются системы Циглера-Натта, образуемые из комплекса переходного металла (Ti, V, Cr, Zr) и алкилалюминия:
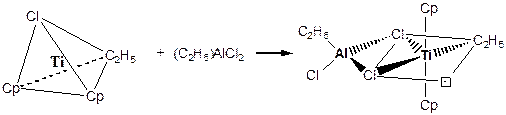 (3.41)
(3.41)
В свободном состоянии тетраэдрический комплекс [Cp2Ni(C2H5)Cl] не имеет вакантных мест и каталитической активностью не обладает. Молекула алкилалюминия ((C2H5)AlCl2) обладает высокой комплексообразующей способностью и координируется с комплексом титана посредством двух мостиковых связей через атомы хлора. При этом координационная сфера титана меняет свою симметрию с тетраэдрической на октаэдрическую и в ней образуется вакантное координационное место, способное принимать олефин. Именно такой комплекс и обладает высокой каталитической активностью в реакции полимеризации.
Высокая скорость стадии b-элиминирования (r2) характерна для комплексов мягких металлов VIII группы (например, Ni и Rh) в низших степенях окисления (0, +1). С позиции концепции ЖМКО это объясняется стремлением перестройки комплекса из энергетически невыгодного состояния (мягкий металл)-(жесткий лиганд-алкил) в более энергетически выгодное - (мягкий металл)-(мягкий лиганд-олефин).
Рассмотрим на примере каталитического цикла димеризации этилена на родиевом комплексе:
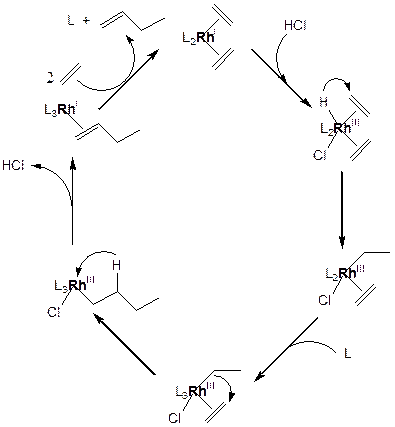 (3.42)
(3.42)
Лимитирующей стадией каталитического цикла является внедрение этилена по связи RhIII–алкил. Образовавшийся s-комплекс имеет так называемую "диссимметрию жесткости-мягкости" (RhIII – относительно мягкий центр, бутил – относительно жесткий центр), поэтому алкильный лиганд легко и быстро подвергается b-элиминированию с образованием мягкого комплекса RhI–олефин.
3.3.4. ДИСПРОПОРЦИОНИРОВАНИЕ (МЕТАТЕЗИС)
Реакция диспропорционирования (метатезис) состоит в обмене алкилиденовыми остатками между двумя молекулами олефинов:
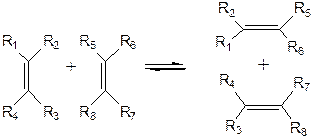 (3.43)
(3.43)
Установлено, что реакция катализируется алкилиденовыми (карбеновыми) комплексами, которые образуются следующим образом:
WVICl6 + C2H5AlCl2 ßà C2H5WVICl5 + AlCl3 (3.44)
C2H5WVICl5 à CH3-CH=WVIHCl5 à CH3-CH=WIVCl4 + HCl
Сначала по обменной реакции с алкилалюминием из хлорида вольфрама (VI) образуется алкильный комплекс, который далее, в результате реакции a-элиминирования, превращается в карбеновый (этилиденовый) комплекс. Затем по реакции восстановительного элиминирования HCl образуется комплекс вольфрама (IV), имеющий вакантное координационное место, способное принять олефин. Это комплекс и является эффективным катализатором диспропорционирования. Каталитический цикл выглядит следующим образом:
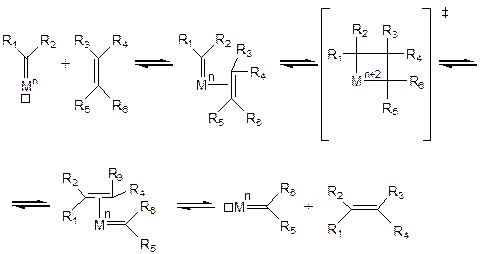 (3.45)
(3.45)
Вначале на вакантное место в комплексе координируется молекула олефина. Затем через переходное состояние, являющееся результатом реакции окислительного сочетания карбенового и олефинового лигандов, образуются новые карбеновый и олефиновый лиганды. Новый олефин затем диссоциирует в раствор.
3.3.5. ОКИСЛЕНИЕ
Существует два основных механизма реакций окисления, катализируемых комплексами переходных металлов: гимолитический и гетеролитический.
3.3.5.1. РАДИКАЛЬНО-ЦЕПНОЕ ОКИСЛЕНИЕ (ГОМОЛИТИЧЕСКИЙ МЕХАНИЗМ)
Радикально-цепное окисление органических соединений катализируется соединениями жестких металлов (например Mn+3, Co+3) в высокой степени оксиления, способных восстанавливаться до степени окисления +(n-1). Механизм таких реакций заключается в образовании радикалов при разрыве связи в органическом реагенте с одновременным восстановлением металла.
В промышленности по такому механизму окисляют, в частности, ацетальдегид до уксусной кислоты, применяя в качестве катализатора органо-растворимые соли Mn+3 или Co+3. Каталитический цикл протекает через следующие стадии:
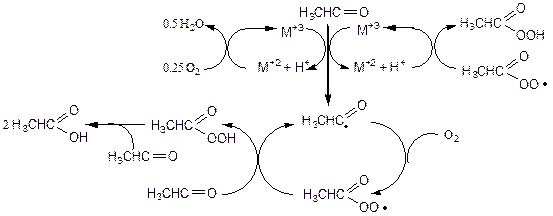 (3.46)
(3.46)
Вначале гомолитически рвется связь С-Н при карбонильном углероде ацетальдегида, давая радикал водорода и ацильный радикал. Атом водорода окисляется катионом металла (М+3) до протона, а металл при этом восстанавливается до М+2. Ацил-радикал присоединяет молекулу кислорода образуя перацетат-радикал, который отрывает атом водорода от молекулы ацетальдегида, давая молекулу надуксусной кислоты и новый ацил-радикал (цикл по ацил-радикалу замыкается). Надуксусная кислота, являясь очень сильным окислителем, окисляет ацетальдегид, в результате чего образуются две молекулы продукта - уксусной кислоты. Каталитический цикл по металлу замыкается окислением М+2 до М+3 либо кислородом, либо перацетат-радикалом.
При наличии соединений меди в качестве сокатализатора протекает реакция образования уксусного ангидрида за счет превращения ацил-радикала в координационной сфере Cu+2:
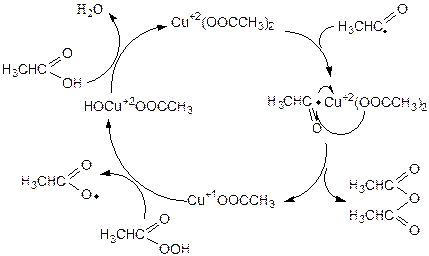 (3.47)
(3.47)
Ацил-радикал входит в координационную сферу ацетата меди (II), где отдает свой электрон иону меди и соединяется с ацетат-анионом, образуя продукт реакции - уксусный ангидрид. Ацетата меди (I) окисляется надуксусной кислотой до основного ацетата меди (II), который на завершающей стадии цикла по обменной реакции с уксусной кислотой снова превращается в ацетата меди (II). В промышленности таким способом осуществляют процесс совместного получения уксусной кислоты и уксусного ангидрида.
3.3.5.2. ГЕТЕРОЛИТИЧЕСКИЙ МЕХАНИЗМ ОКИСЛЕНИЯ
Окисление органических соединений по гетеролитическому механизму осуществляется внутри координационной сферы комплекса металла. Примерами промышленно важных процессов, протекающих по такому механизму являются эпоксидирование непредельных соединений органическими гидропероксидами и окисление этилена до ацетальдегида.
3.3.5.2.1. Эпоксидирование олефинов органическими гидроперекисями
Реакцию эпоксидирования олефинов органическими гидропероксидами:
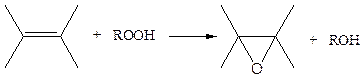 (3.48)
(3.48)
катализируют комплексы жестких металлов: Mo, W, Ti в высоких степенях окисления (+4) - (+6).
Ключевая стадия каталитического цикла – активация гидропероксида в координационной сфере металла за счет передачи электронной плотности от атома кислорода гидроперексида к металлу, в результате чего один из атомов кислорода приобретает электрофильные свойства и способность взаимодействовать с p-электронами олефина, образуя эпоксид:
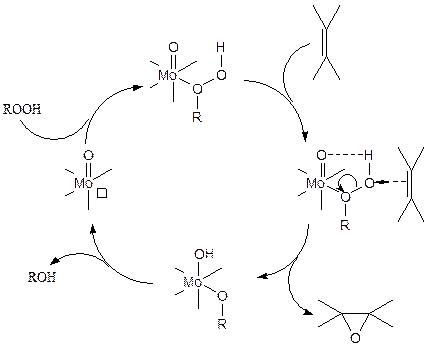 (3.49)
(3.49)
Факторы, влияющие на эффективность катализаторов эпоксидирования.
Увеличение льюисовской кислотности металла (жесткости) увеличивает эффективность катализа. Например, комплексы MoVI активнее комплексов WVI.
Электронно-акцепторные лиганды также увеличивают эффективность катализатора. Например, ацетилацетонатный комплекс молибдена [MoO2(acac)2] активнее гликолевого комплекса [MoO2(diol)2] из-за сильных электроно-акцепторных свойств ацетилацетона (обусловленных обратным p-связыванием с p*-разрыхляющими орбиталями карбонильных групп.
3.3.5.2.2. Окисление этилена до ацетальдегида
Процесс окисления этилена до ацетальдегида проводят в присутствии каталитической системы, представляющей собой смесь хлоридов PdII/CuII. В основе процесса лежит стехиометрическая реакция окисления этилена комплексом PdII, которая протекает по следующему механизму:
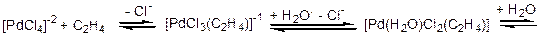 (3.50)
(3.50)
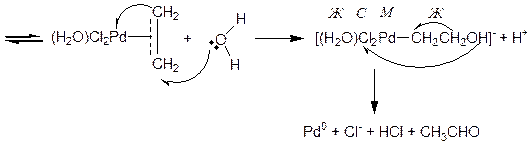
Анионный плоскоквадратный комплекс [PdCl4]- последовательно обменивает один Cl--лиганд на этилен, а второй на молекулу воды (реакция протекает в водном растворе соляной кислоты). Н2О, в отличие от Cl-, не образует дополнительных донорных p-связей с орбиталями палладия, делая тем самым этилен более доступным для внешней нуклеофильной атаки молекулой воды из раствора. В результате взаимодействия с водой образуется оксиэтильный лиганд. Образовавшийся комплекс обладает сильной ассиметрией жесткости/мягкости, так как мягкий центральный атом окружен жесткими и средне-жесткими лигандами (на схеме 3.50 они обозначены соответствующими начальными буквами), поэтому он быстро распадается с восстановлением Pd2+ до Pd0 и окислением оксиэтильного лиганда до продукта реакции - ацетальдегида.
В присутствии ионов Cu2+ протекает быстрая окислительно-восстановительная реакция приводящая к регенерации активной формы палладия:
Pd0 + 2Cu+2 à Pd+2 + 2Cu+ (3.51)
Одновалентные ионы меди в кислой среде, в свою очередь, легко окисляются кислородом:
2Cu+ + 0,5O2 + 2H+ à H2O + 2Cu2+ (3.52)
Таким образом, можно сказать, что каталитический цикл в данном процессе состоит из последовательности некаталитических окислительно-восстановительных реакций, приводящей к образованию ацетальдегида и регенерации исходной формы соединений палладия и меди.
3.3.6. ПРИСОЕДИНЕНИЕ ПРОТОНОДОНОРНЫХ ВЕЩЕСТВ К ОЛЕФИНАМ И АЦЕТИЛЕНАМ
Комплексы переходных металлов (таких, как Hg(II), Cu(I), Zn(II), Cd(II), Rh(II), Pd(II)) катализируют присоединение протонодонорных веществ (НХ) к олефинам и ацетиленам. Общая схема катализа:
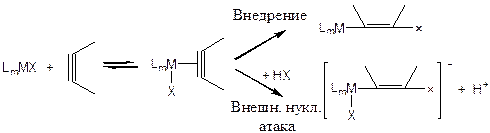 (3.53)
(3.53)
Механизм включает в себя стадию координации непредельного соединения в комплекс металла. Далее превращение может протекать либо по механизму внедрения непредельного лиганда по связи М-Х, либо по механизму внешней нуклеофильной атаки на непредельный лиганд анионом Х-. Затем происходит образование продукта и регенерация катализатора в результате внешней электрофильной атаки протоном:
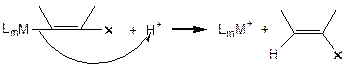 (3.54)
(3.54)
Аналогично протекает реакция и для олефинов. Особенно эффективен катализ комплексами мягких переходных металлов реакции присоединения мягких (слабых) кислот. Примером может служить процесс получения адиподинитрила:
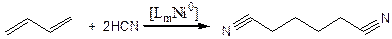 (3.55)
(3.55)
3.3.7. СИНТЕЗЫ НА ОСНОВЕ ОКИСИ УГЛЕРОДА
Из реакций окиси углерода, катализируемых гомогенными комплексами переходных металлов, промышленное значение имеют: карбонилирование метанола, карбоксилирование ацетилена и олефинов и гидроформилирование олефинов.
3.3.7.1. Карбонилирование метанола с получением уксусной кислоты
Реакция карбонилирования метанола протекает по следующему суммарному уравнению:
CH3OH + CO à CH3COOH (3.56)
Катализатором данной реакции служит анионный комплекс родия [RhI(CO)2I2]-, который образуется в реакционной массе из RhCl3 и HI под давлением СО. Реакция протекает в присутствии промотора СН3I.
Каталитический цикл:
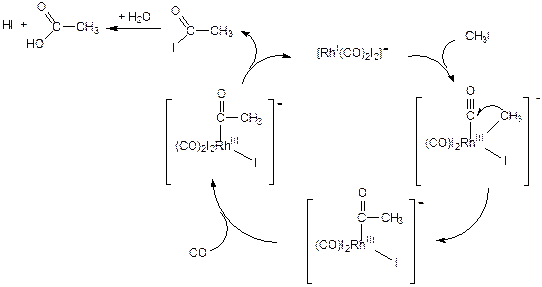 (3.57)
(3.57)
Каталитический цикл начинается со стадии окислительного присоединения йодистого метила к комплексу родия. Следующей стадией является реакция внедрения СО по связи Rh-СН3, приводящая к образованию ацильного лиганда и освобождению вакансии в координационной сфере. Вакантное координационное место занимает молекула СО. Каталитический цикл замыкается стадией восстановительного элиминирования йодистого ацила с регенерацией исходной формы катализатора. Продукт реакции - уксусная кислота - образуется по реакции гидролиза йодангидрида. Регенерация промотора протекает по реакции нуклеофильного замещения:
HI + CH3OH à CH3I + H2O (3.58)
Интересно заметить, что в каталитическом цикле нет стадии прямого взаимодействия метанола с СО.
3.3.7.2. Карбоксилирование непредельных соединений
Комплексы переходных металлов, из которых наиболее активными являются Со, Pd, Rh, катализируют реакции карбоксилирования непредельных соединений с получением карбоновых кислот и их производных:
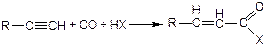 (3.59)
(3.59)
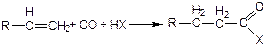 (3.60)
(3.60)
Каталитический цикл на примере реакции карбоксилирования этилена в водной среде выглядит следующим образом:
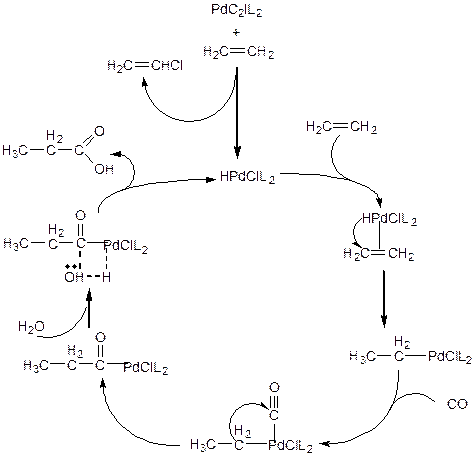 (3.61)
(3.61)
Сначала образуется каталитически активный гидридный комплекс палладия по реакции гетеролитического присоединения. Далее следует, собственно, каталитический цикл, начинающийся с присоединения этилена на вакантное координационное место. Затем протекает реакция внедрения этилена по связи Pd-H. На освободившееся координационное место присоединяется молекула СО, которая затем внедряется по связи Pd-C2H5 с образованием пропионильного лиганда. Следующая, завершающая цикл стадия, может рассматриваться как гетеролитическое присоединение гидрида с внешней нуклеофильной атакой молекулой воды на карбонильный углерод пропионила. На этой стадии в раствор выходит продукт реакции - пропионовая кислота и регенерируется исходная форма катализатора.
В зависимости от природы протонодонорного реагента (НХ в реакцииях (3.59), (3.60)), вступающего в реакцию на последней стадии каталитического цикла (3.61), реакция карбоксилирования приводит к образованию различных продуктов:
| НХ: | H2O | ROH | RNH2 | RCOOH |
| Продукт: | Кислоты | Эфиры | Амиды | Ангидриды |
Практическое значение для промышленного органического синтеза имеют следующие две реакции карбоксилирования ацетиленовых углеводородов, катализируемые тетракарбонилом никеля(0).
Карбонилирование ацетилена в водной среде приводит к образованию акриловой кислоты (способ, альтернативный гетерогенно-каталитическому окислению акролеина):
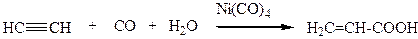 (3.62)
(3.62)
Карбонилирование метилацетилена в водно-метанольном растворе с образованием метилметакрилата (способ, альтернативный кислотно-каталитическому получению из ацетонциангидрина):
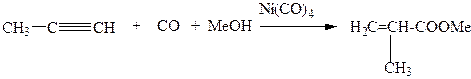 (3.63)
(3.63)
3.3.7.3. Гидроформилирование алкенов с получением альдегидов
Гидроформилирование a-олефинов приводит к образованию альдегидов, имеющих на один атом углерода больше, чем исходный олефин. Альдегиды получаются как линейного, так и разветвленного строения:
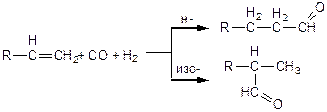 (3.64)
(3.64)
В промышленном синтезе находят применение в качестве катализаторов, гидридные комплексы только двух металлов - CoI и RhI - модифицированные различными лигандами.
Рассмотрим механизм катализа на примере комплекса HCoI(CO)4:
(3.65)
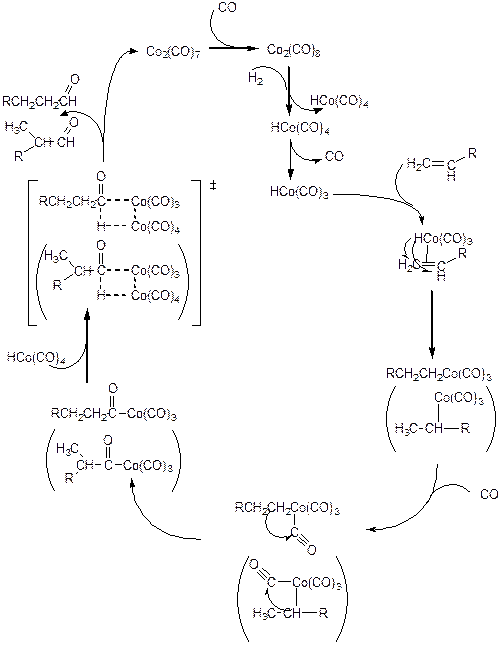
Образование каталитически активного гидридного комплекса происходит по реакции гомолитического присоединения водорода к октакарбонилу дикобальта Сo02(CO)8. Образовавшийся HCoI(CO)4 является координационно насыщенным, поэтому для продолжения каталитического цикла необходима диссоциация одной молекулы СО. На освободившееся координационное место присоединяется олефин. Далее следует внедрение олефина по связи Со-Н с образованием алкильного лиганда. Внедрение может протекать как по правилу Марковникова, так и против в зависимости от природы комплекса (п. 3.3.2). При этом образуется либо вторичный, либо первичный алкил, соответственно. Затем следуют стадии координирования еще одной молекулы СО и внедрение СО по связи металл-алкил. При этом образуется ацильный лиганд либо линейного, либо разветвленного строения. Образовавшийся комплекс взаимодействует далее с комплексом HCoI(CO)4 по реакции, обратной гомолитическому присоединению, в результате чего образуется продукт реакции - альдегид и би-ядерный комплекс Сo02(CO)7. Последний присоединяет молекулу СО, и на этом каталитический цикл завершается.
Активность комплексов родия в реакциях гидроформилирования в 100-10000 раз выше, чем у кобальта. С позиций концепции ЖМКО это можно объяснить тем, что более мягкий RhI гораздо легче координирует мягкий лиганд - олефин, а также, тем, что стадия образования продукта протекает гораздо быстрее, так как при этом устраняется ассиметрия жесткости/мягкости между мягким центром (RhI) и жестким ацильным лигандом.
4. ПРОМЫШЛЕННЫЕ ПРОЦЕССЫ МЕТАЛЛОКОМПЛЕКСНОГО КАТАЛИЗА
Спектр промышленных процессов, катализируемых комплексами переходных металлов очень широк и иллюстрируется следующей диаграммой:

Рис. 4.1. Промышленные процессы, катализируемые комплексами переходных металлов.
Несмотря на то, что доля гомогенного комплексного катализа оценивается в 10-15% от всех промышленных процессов органического синтеза, тем не менее ряд крупнотоннажных процессов проводится в присутствии именно таких катализаторов.
Годовая мощность некоторых типов процессов составляет (в млн. тонн в год):
- Окисление – 14,0
- Реакции СО – 8,0
- Гидрирование – 1,4
- Олигомеризация - 0,8
- Гидроцианирование – 0,4
4.1. ОСОБЕННОСТИ ТЕХНОЛОГИИ ПРОЦЕССОВ МЕТАЛЛОКОМПЛЕКСНОГО КАТАЛИЗА
Из содержания предыдущей главы (гл. 3) видно, что одной из особенностей процессов, катализируемых комплексами переходных металлов, является то, что реагенты представляют собой малые молекулы, и, чаще всего, газы (СО, Н2, этилен, пропилен, метанол). Это заставляет применять высокие давления (сотни атмосфер) на стадии синтеза для обеспечения высоких концентраций газов-реагентов в жидкой реакционной массе и стойкости комплексных соединений (катализаторов).
Также, по причине газообразного состояния реагентов, в качестве реакторов используют, как правило, барботажные колонны и реактора с мешалкой (Рис. 2.5, аппараты 1, 3, 8), которые обеспечивают эффективный контакт газовой и жидкой фаз с целью учкорения массообменных процессов.
Во многих процессах на стадии синтеза или регенерации катализаторов используются агрессивные среды, что заставляет изготавливать аппаратуру из дорогих кислото- и коррозионно-стойких материалов.
Одной из самых важных особенностей металлокомплексных катализаторов является высокая цена на образующие их металлы (Табл. 4.1). Поэтому, экономика производства заставляет организовывать сложные узлы рециркуляции катализаторов для обеспечения минимальных потерь дорогостоящих металлов.
Табл. 4.1.
Ориентировочные цены на металлы (по данным лондонской биржи) на октябрь месяц 2003 года.
| № в порядке убывания цены | Металл | Цена, US$/кг |
| 1 2 3 4 5 6 7 8 9 10 11 12 13 14 | Pd Pt Rh Au Ir Ru Ag Co Mo Ni Ti Cu Mn W (в виде WO3) | 38 870 20 318 16 500 13 816 2 050 1 060 187 21,5 13 10 5.5 1.8 1.2 0.042 |
Ниже подробно рассмотрены некоторые, наиболее важные, промышленные процессы металлокомплексного катализа.
4.2. ОКСОСИНТЕЗ
Процесс оксосинтеза (гидроформилирования олефинов) протекает по суммарной реакции:
R-CH=CH2 + CO + H2 à R-CH2-CH2-CH=O + R-CH(CH3)-CH=O (4.1)
и направлен на получения линейных альдегидов, служащих сырьем для получения линейных первичных спиртов по реакции гидрирования.
В настоящее время в промышленности используют четыре типа катализаторов на основе комплексов Со и Rh, различающихся лигандным окружением:
1. HCo(CO)4 - тетра-карбонил кобальт гидрид
2. HCo(CO)3PAlk3 - триалкилфосфин-три-карбонил кобальт гидрид
3. HRh(CO)(PPh3)3 - три-(трифенилфосфин)-карбонил родий гидрид
4. HRh(CO)(TPPTS)3 - три-(трифенилфосфинтрисульфат)-карбонил родий гидрид (ТРРТS = P(PhSO3Na)3).