
Влияние природы атакующего лиганда ( Y - ) на скорость реакции.








Поскольку внедрение протекает через стадию нуклеофильной атаки, то реакционная способность растет с ростом электроно-донорного характера лиганда Y-. Так например, в реакции карбонилирования, протекающей по реакции, аналогичной (3.25) и катализируемой комплексом [R–MnI(CO)5], реакционная способность падает в ряду:
R: n-C3H7 > Et > Ph > Me >> CH2Ph, CF3
Влияние природы металла.
В группах d-элементов с увеличением жесткости металла (т.е. в направлении уменьшения порядкового номера в группах периодической системы) увеличивается реакционная способность комплекса:
Co > Rh > Ir; Mn > Re; Cr > Mo > W
Это влияние объясняется повышением электроноакцепторных свойств металлов, что приводит к большему смещению электронной плотности по связи металл-Х в сторону металла, и, как следствие, к увеличению электрофильности лиганда Х, что облегчает атаку на него лиганда Y-.
3.2.5. a - И b -ЭЛИМИНИРОВАНИЕ
s-связанные органические лиганды могут отщеплять и передавать металлу анионы (H-, Cl-, AcO- и др.). Такие реакции могут протекать только в координационно-ненасыщенных комплексах, т.к. отщепляемый анион должен занять свободное координационное место.
b -элиминированием – называют реакции, в которых анион отщепляется от b-атома лиганда. Фактически, это реакция – обратная реакции внедрения и также протекает через циклическое переходное состояние:
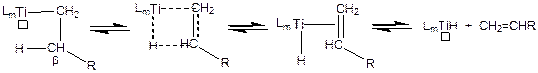 (3.26)
(3.26)
Уравнением 3.26 представлена реакция b-элиминирования гидрид-аниона от алкильного лиганда с образованием олефина, который затем диссоциирует из комплекса в раствор. Пустым квадратом обозначено свободное координационное место в координационной сфере комплекса титана.
a -элиминированием – называют реакции, в которых анион отщепляется от a-атома лиганда. a-элиминирование наиболее характерно для комплексов W и Mo.
Отрыв Н- от a-атома углерода лиганда приводит к образованию карбенов:
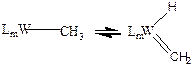 (3.27)
(3.27)
Даже при наличии у лиганда как a-, так и b-атомов, способных к элиминированию, в комплексах вольфрама и молибдена предпочтительнее протекает a-элиминирование:
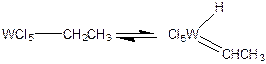 (3.28)
(3.28)
3.2.6. ВНЕШНЯЯ НУКЛЕОФИЛЬНАЯ И ЭЛЕКТРОФИЛЬНАЯ АТАКА
При внешней нуклеофильной атаке происходит взаимодействие лиганда с нуклеофилом, находящимся в растворе (вне комплекса). Нуклеофилами чаще всего выступают следующие соединения: HO-, RO-, R3N, H-, CH3-, H2O и др. В качестве лигандов, подвергающихся внешней нуклеофильной атаке, чаще всего выступает СО, олефины, ацетилены, нитрилы, т.е. молекулы, содержащие кратную связь.
Практически важной реакцией внешней нуклеофильной атаки является алкоксильная атака на карбонильные комплексы, приводящая к образованию алкоксикарбонильного комплекса. Она характерна для комплексов металлов VII и VIII групп (Mn, Re, Fe, Rh, Os, Co, Ru, Ir, Pd, Pt и Hg):
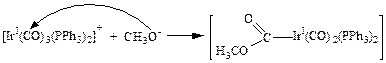 (3.29)
(3.29)
Внешней электрофильной атаке подвергаются чаще всего комплексы олефинов и ароматических соединений. Нуклеофильный или электрофильный характер поведения этих лигандов зависит от соотношения s/p-хатактера координационной связи.
Если преобладает обратное p-связывание, то электронная плотность смещается от металла к олефину. В этом случае легко протекает атака электрофилом. Электорофильной атаке на лиганды также способствуют низшие степени окисления металла (0, +1) и анионные комплексы. И напротив, высокие степени окисления металла (+2, +3 и выше) и катионные комплексы способствуют внешней нуклеофильной атаке.
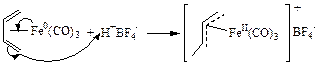 (3.30)
(3.30)
В реакции (3.30) протон атакует диеновый лиганд, что приводит к образованию p-аллильного комплекса.
В следующем примере (3.31) карбокатион отрывает гидрид-анионы от координированного в комплекс циклогексадиена с образованием ароматического комплекса:
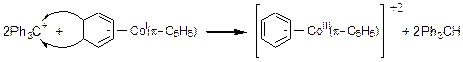 (3.31)
(3.31)
Практически интересным случаем взаимодействия лигандов с внешним электрофилом является взаимодействие атома кислорода координированной молекулы СО с сильными кислотами Льюиса или протоном. Такая двойная активация СО приводит к сильному ослаблению связи С-О и ускорению реакции внедрения карбонила:
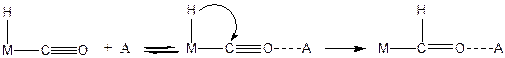 (3.32)
(3.32)
Поэтому добавление кислот Льюиса к карбонильным комплексам металлов, может быть перспективным для катализа реакции Фишера-Тропша.
5.3. МЕХАНИЗМЫ РЕАКЦИЙ, КАТАЛИЗИРУЕМЫХ КОМПЛЕКСАМИ ПЕРЕХОДНЫХ МЕТАЛЛОВ
3.3.1. ГИДРИРОВАНИЕ
Комплексы практически всех переходных металлов проявляют каталитическую активность в реакциях гидрирования. Но наибольшей активность отличаются комплексы металлов VIII группы. Они эффективно катализируют присоединение водорода по кратным связям (C=C, CºC, C=O, CºN) в достаточно мягких условиях. Подбором металла и его лигандного окружения можно добиваться высокой селективности гидрирования (например, селективнаое гидрирование связи CºC до C=C; или гидрирование C=C в непредельных альдегидах не затрагивая связь C=O и пр.).
Для протекания реакции гидрирования водород должен войти в координационную сферу металла. Введение водорода может протекать по трем рассмотренным ранее механизмам: гомолитическое, гетеролитическое и окислительное присоединение.
Например, для комплекса Вилкинсона RhI(PPh3)3Cl (комплекс родия d8 квадратного строения) характерно окислительное присоединене водорода, сопровождающееся последующей диссоциацией одного трифенилфосфинового лиганда:
16e 18e 16e
RhIL3Cl + Н2 ßà Н2RhIIIL3Cl ßà Н2RhIIIL2Cl + L (3.33)
Образующийся в результате 16-электронный координационно-ненасыщенный гидридный комплекс является активным катализатором реакции гидрирования. После этого следует каталитический цикл (3.34), который проходит через стадии: присоединения олефина, внедрения олефина по связи Rh-H, восстановительного элиминирования продукта гидрирования, и, снова, окислительного присоединения водорода (регенерация катализатора).
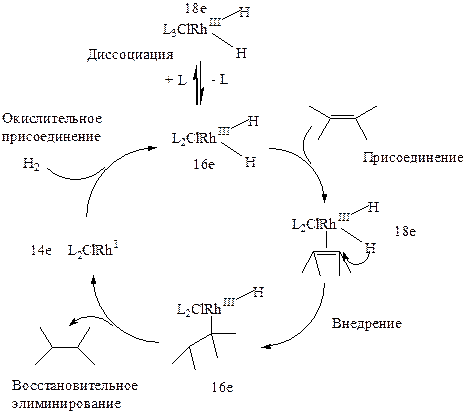 (3.34)
(3.34)